
06. tbl. 97. árg. 2011
Fræðigrein
Fenýlketónúría á Íslandi
Ágrip
Inngangur: Fenýlketónúría (PKU) er efnaskiptasjúkdómur sem orsakast af stökkbreytingu í fenýlalanín-hýdroxýlkljúfs (PAH) geninu. Skimun fyrir PKU hófst árið 1972 á Íslandi. Amínósýrunni fenýlalaníni (Phe) er breytt í týrósín (Tyr) fyrir tilstuðlan PAH ásamt hjálparþættinum tetrahydrobiopterín (BH4). Uppsöfnun á Phe veldur þroskaskerðingu og flogum. Meðferð PKU er Phe-skert fæði ásamt nýrri aðferðum eins og BH4-gjöf í stórum skömmtum. Markmið rannsóknarinnar var að draga saman upplýsingar um sjúkdóminn, árangur kembileitar og meðferðar. Einnig var BH4-meðferð athuguð.
Efniviður og aðferðir: Rannsóknin var afturskyggn þar sem upplýsingar voru fengnar úr sjúkraskrám Landspítala. Athuguð voru Serum-Phe (S-Phe) gildi, aldur við upphaf meðferðar, gerðir stökkbreytinga sem og núverandi meðferð. Skoðað var BH4-hleðslupróf hjá fjórum einstaklingum.
Niðurstöður: Frá árinu 1947 hafa 27 greinst með PKU hér á landi. Nýgengi árin 1972-2008 er 1/8400 lifandi fæddum. Klassísk PKU er algengust á Íslandi. Einstaklingar greindir eftir að kembileit hófst hafa allir eðlilega greind. Aldur við upphaf meðferðar og S-Phe gildi lækka þegar líður á. Fundist hafa 12 stökkbreytingar hérlendis. Séríslenska stökkbreytingin Y377fsdelT svarar ekki BH4-hleðsluprófi. Tveir einstaklingar með PKU eru nú á BH4-meðferð og fjórir til viðbótar gætu svarað BH4-hleðsluprófi.
Ályktun: Nýgengi PKU virðist aðeins hærra hér en í nágrannalöndunum. Meðferð hér gengur vel og fylgir alþjóðlegum markmiðum. Kembileit er örugg og hefur skilað tilsettum árangri. BH4-gjöf er meðferðarkostur sem gæti komið fleirum með PKU til góða.
Inngangur
Fenýlketónúría (PKU) er algengasti meðfæddi efnaskiptagalli sem þekktur er á Íslandi. Kembileit hófst árið 1972 með þunnlagsskilju. PKU er sjálflitningsvíkjandi erfður efnaskiptasjúkdómur. Á Íslandi höfðu fundist níu gerðir stökkbreytinga árið 1997, algengust af þeim var Y377fsdelT sem er séríslensk.1
Í PKU er ensímvirkni fenýlalanín-hýdroxýlkljúfs (PAH) gölluð vegna stökkbreytinga í PAH-geninu sem er á langa armi litnings númer 12.2 Fundist hafa yfir 500 stökkbreytingar og valda þær mismikilli virkniskerðingu á PAH. Þessi ensímgalli veldur uppsöfnun á amínósýrunni fenýlalanín (Phe) í líkamanum og þar af leiðandi hækkuðu Serum-fenýlalaníni (S-Phe). PAH sér um fyrsta skrefið í að breyta Phe í amínósýruna týrósín (Tyr).2 Þeir þættir sem stjórna virkni PAH eru Phe, hjálparþátturinn tetrahydrobiopterin (BH4) og afturkræf fosfórun.3
PKU er skipt í fjóra flokka eftir alvarleika og gerð þar sem klassísk PKU er alvarlegust.4 Sjúkdómsmynd PKU er mikil þroskaskerðing ásamt vaxtarskerðingu og flogum. Án meðferðar búa sjúklingar við mikla andlega fötlun. Sýnd sjúkdómseinkenna er afar mismunandi og virðist ekki fara beint eftir tegundum stökkbreytinga eða S-Phe gildum.5 Þegar uppsöfnun verður, aukast aðrar afleiður Phe. Í upphafi var talið að afurðir þessara hliðarferla eins og phenylpyruvic-sýra væru helsti orsakavaldur sjúkdómsmyndarinnar.3 Seinna hefur komið í ljós að sjálf uppsöfnunin á Phe í miðtaugakerfinu (MTK) er stærsti þáttur meingerðar PKU.6 Phe-uppsöfnun í MTK veldur hindrun á taugaslíðringu sem og aftaugaslíðrun og þannig má útskýra sjúkdómsmyndina.7
Hefðbundin meðferð PKU í dag er Phe-skert fæði þar sem reiknaður er út hámarksdagskammtur Phe og meðferð fylgt eftir með blóðgildum. Gefin eru aukalega ýmis vítamín, snefilefni og prótein sem ekki fást í skerta fæðinu. Því nær eðlilegum mörkum (85-170 µM/L) sem S-Phe eru í æsku og því fyrr sem meðferð hefst, verður vitsmunaleg geta einstaklingsins betri.8, 9 Í ljós hefur komið að meðferð fyrir lífstíð er mikilvæg til að halda eðlilegri andlegri getu.10, 11 Margar nýjar meðferðir hafa verið reyndar á undanförnum árum. BH4-gjöf um munn í mjög stórum skömmtum hefur gefist vel á stökkbreytingar þar sem einhver PAH-virkni er til staðar.12 Einnig er farið að gefa stóru hlutlausu amínósýruna lýsín í töfluformi (PreKUnil‰) en hún keppir við Phe um inngöngu í MTK yfir heilablóðstálma. Veita þessar meðferðir einstaklingunum mun meira frelsi í hefðbundnu með-ferðinni.
Tilgangur rannsóknarinnar var að afla nánari upplýsinga um PKU á Íslandi og bæta við áður útgefið efni.1 Markmið rannsóknarinnar var þríþætt. Í fyrsta lagi að finna nákvæmt nýgengi PKU frá því að kembileit hófst 1972 til 2008. Í öðru lagi að meta gæði meðferðar og kembileitar og í þriðja lagi að lýsa gerðum og fjölda stökkbreytinga. Að lokum er samantekt um virkni hjálparþáttarins BH4 um munn.
Efniviður og aðferðir
Rannsóknin var afturskyggn. Upplýsingar voru fengnar úr sjúkraskám og frá rannsóknarstofu Landspítala. Skráð voru S-Phe gildi, aldur við upphaf meðferðar, form og fyrirkomulag núverandi meðferðar, andleg geta og hugsanlegir fylgikvillar til mats á gæðum og árangri meðferðar. Í rannsókninni voru allir einstaklingar greindir með PKU á Íslandi frá 1947 til 2008 með þunnlagsskiljuaðferð.13 Notaðar voru upplýsingar um fjölda fæðinga á tímabilunum 1947-1972 og 1972-2008 frá fæðingaskrá og Hagstofu Íslands við útreikninga á nýgengi. Reiknuð voru meðaltöl og miðgildi og borin saman í tímaröð. Athugaðar voru gerðir stökkbreytinga í PAH-geninu. Blóðsýni PKU-einstaklinga sem fæddir voru 2006 og seinna voru send til Kennedy Institute í Kaupmannahöfn og stökkbreytingar í PAH-geni greindar með keðjumögnun (PCR) og 13 pörum af þreifurum (primers) fyrir ríbósakjarnasýru (RNA) splæsingu.1 Stökkbreytingagerðir eldri einstaklinga voru þekktar.
Athugað var hvort séríslensk stökkbreyting, Y377fsdelT, ásamt Y414C svöruðu BH4 hleðsluprófi. Var það hluti af almennri meðferð sjúklinga en ekki hluti rannsóknar. Við hleðslupróf er einstaklingi gefið BH4 degi eftir að hann hefur safnað Phe í líkama með eðlilegu fæði og S-Phe gildi mæld reglulega eftir inntöku.14
Beitt var lýsandi tölfræði. Tölvuforritið Excel var notað við tölfræðilega og myndræna úrvinnslu.
Fengið var leyfi fyrir rannsókninni frá Siðanefnd Landspítalans og Persónuvernd. Einnig lá fyrir heimild lækningaforstjóra til rannsóknarframkvæmdar á spítalanum.
Niðurstöður
Á Íslandi hafa 27 manns greinst með PKU frá 1947, 18 karlar og 9 konur. Frá því kembileitin hófst árið 1972 til loka ársins 2008 greindust 19 með efnaskiptagallann og gefur það nýgengi 1/8400 lifandi fæddra. Ekki er vitað um falskt neikvætt tilfelli á því tímabili. Frá 1947 til 1972 greindust níu manns með PKU, eða 1/14000 fæddum.
Allir þeir sem greinst hafa með PKU eftir að kembileit hófst eru heilbrigðir. Af átta manns sem greindust áður en kembileitin hófst eru sex andlega fatlaðir. Einnig má sjá að meðferð hefst mun fyrr eftir að kembileit hófst (tafla I).
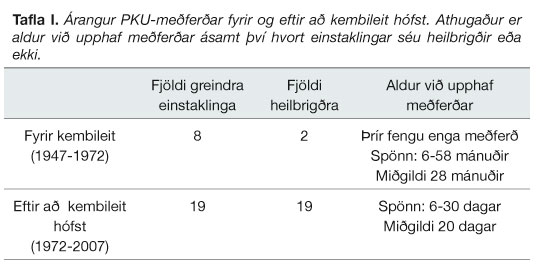{kind=link}
Alvarlegasti flokkurinn, klassísk PKU, er algengastur á Íslandi (tafla II).
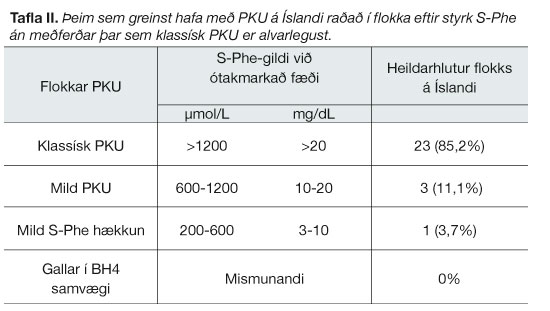{kind=link}
Allir nema einn greindir eftir 1972 eru á meðferð til að halda S-Phe gildum niðri. Enginn þeirra er andlega fatlaður eða með þekkta fylgikvilla. Eftir að kembileit hófst hefja sjúklingar meðferð við yngri aldur (mynd 1).
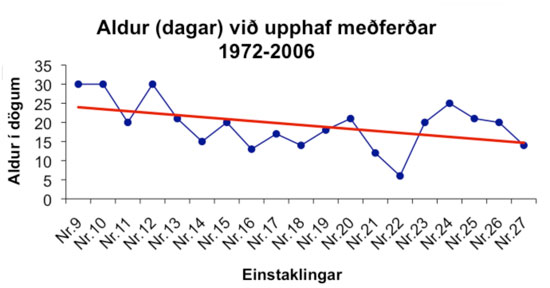{kind=link}
Frá 1989 til 2002 lækkuðu S-Phe-gildi að meðaltali um fjórðung fyrstu 42 mánuði meðferðar (mynd 2). Fyrsta mánuði var sleppt því engin meðferð er í fyrri hluta hans.
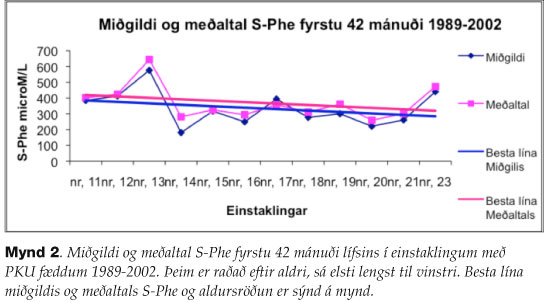{kind=link}
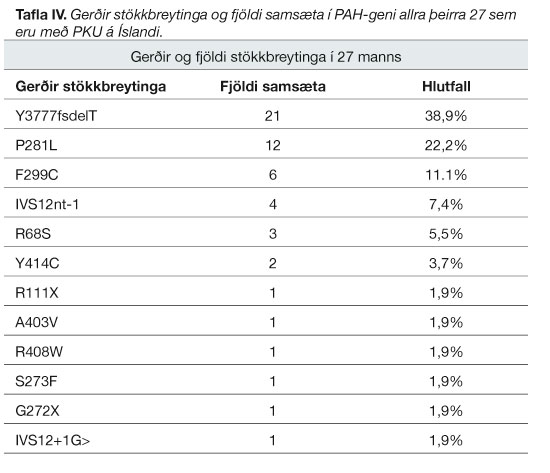
Stökkbreytingagerð í PAH-geninu hefur verið athuguð hjá öllum einstaklingum greindum með PKU á Íslandi. Greinst hafa 12 gerðir stökkbreytinga hér á landi og 54 samsætur. Þrjár stökkbreytingar eru í rúmlega 2/3 samsæta, Y377fsdelT, P281L og F299C. Er íslenska stökkbreytingin Y377fsdelT algengust þeirra (tafla IV).
{kind=link}
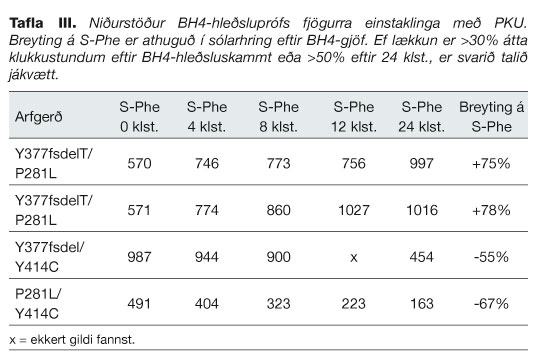
Tveir einstaklingar með stökkbreytinguna Y377fsdelT á annarri PAH-samsætunni og P281L á hinni, gengust undir BH4 hleðslupróf sem þeir svöruðu ekki (tafla III). Tveir aðrir með stökkbreytinguna Y414C á annarri samsætunni fóru í hleðslupróf sem þeir svöruðu (tafla III).
{kind=link}
Umræður
Átta einstaklingar fæddir frá 1947 til 1972 greindust með PKU á rannsóknarskeiðinu, margir löngu eftir fæðingu.1 Nýgengi á þessu tímabili er 1/14.000 fæðingum. Líklegt er að einhverjir hafi ekki greinst og dáið ungir í ljósi aðbúnaðar fatlaðra einstaklinga á þessum tíma.
Nýgengi PKU eftir að kembileit hófst árið 1972 og til 2008 er 1/8400 fæðingum. Ekki eru þekkt falskt neikvæð tilfelli og ólíklegt er að einhver séu óuppgötvuð. Samkvæmt Hardy-Weinberg-jafnvæginu er um það bil einn af hverjum 50 Íslendingum sem ber PKU-orsakandi stökkbreytingar. Nýgengi PKU meðal hvítra Vesturlandabúa er almennt um 1/10.000 fæðingum. Er nýgengið hér á landi lægra en á til dæmis Írlandi eða Tyrklandi.15, 16 Rannsóknarskýrsla Árna Þórs Árnasonar læknis árið 2004 leiddi í ljós nýgengi PKU 1/6607 fæðingum á tímabilinu 1984-2003. Áður höfðu Guldberg og fleiri reiknað nýgengi PKU á Íslandi frá 1972-1997 sem 1/10000 fæðingum.1 Ætla má að nýgengið 1/8400 sé réttara vegna lengra rannsóknartímabils.
Efnaskiptagallinn PKU á Íslandi uppfyllir öll skilyrði til kembileitar: a) há tíðni; b) afleiðingar sjúkdómsins slæmar; c) kembileitin örugg og ódýr; d) sjúkdómurinn meðhöndlanlegur á viðráðanlegu verði. Eftir að kembileit hófst árið 1972 byrjar meðferð mun fyrr en áður og hefur enginn hlotið alvarlegan skaða á miðtaugakerfi (tafla I). Allir greindir eftir 1972 eru með eðlilega greind og eru þeir fullgildir og virkir þátttakendur í samfélaginu. Meðferð hefst fyrr eftir því sem líður á rannsóknartímabilið (mynd 1) og er það í samræmi við breyttar alþjóðlegar kröfur í PKU-meðferð. Því fyrr sem meðferð hefst, því betri verður útkoman.17 Í byrjun árs 2008 tók raðmassagreining (tandem mass spectrometry, TMS) við af þunnlagsskiljuaðferð á S-Phe mælingum. Úrvinnsla kembileitarblóðsýna er mun fljótari með TMS og hefst meðferð vonandi fyrr í framhaldinu.
Efnaskiptagallinn PKU er fullkomlega meðhöndlanlegur en með mjög krefjandi meðferð. Þrátt fyrir að einstaklingar njóti góðrar meðferðar á yngri árum glíma margir við ýmsa kvilla seinna á lífsleiðinni, sé meðferð hætt. Má þar nefna þunglyndi, ofvirkni og félagsfælni. Með meðferð alla ævi er hægt að komast hjá síð-komnum fylgikvillum að mestu.18, 19 Hér á landi er lögð mikil áhersla á ævilanga meðferð og allir fæddir eftir 1975 eru á meðferð.
Til nánara mats á árangri meðferðar voru S-Phe gildi skoðuð. Ákveðið var að athuga aðeins fyrstu 42 mánuðina þegar mælingar eru þéttastar. S-Phe-gildi lækkuðu að meðaltali um fjórðung frá 1989 til 2002 (mynd 2) sem er í samræmi við auknar meðferðarkröfur undanfarin ár.10 Fyrstu 8-10 árin miðast meðferð við að halda S-Phe undir 360 µM/L og ekki hærra en 900 µM/L út unglingsárin. Stefnt er á að halda blóðgildum undir 1200 µM/L á fullorðinsárum. Styrkur yfir 1200 µM/L getur valdið bráðri aftaugaslíðringu í miðtaugakerfi.22 Mikill munur er á fjölda mælinga milli einstaklinga og verður munurinn meiri eftir því sem líður á rannsóknartímabilið. Helsti gallinn við að meta PKU-meðferð út frá S-Phe-mælingum er að gildi breytast á tiltölulega stuttum tíma. Hægt er að lækka gildi með hertri meðferð fáum dögum fyrir mælingu og þannig gæti S-Phe ekki endilega endurspeglað meðferðarheldni að fullu. Einnig geta einföld veikindi eða hiti hækkað S-Phe hjá þeim sem eru með PKU. Höfundum er ekki kunnugt um sambærilega athugun á gæðum PKU-meðferðar.
Einn af veikleikum rannsóknarinnar er það voru ekki gerð nákvæm taugasálfræðileg próf. Athuga þarf hvort ástæða sé til að hefja markviss próf á þeim sem greinst hafa með PKU.
18 karlar og 9 konur eru greind með PKU á Íslandi. Þessi kynjamunur er líklega vegna lítils þýðis, almennt eru kynjahlutföll jöfn. PKU hefur ekki áhrif á frjósemi en konur með PKU þurfa að skipuleggja þungun mjög vel og vera í þéttu eftirliti. S-Phe-gildi þurfa að vera innan eðlilegra marka fyrir og á meðgöngu til að koma í veg fyrir skaða á fóstri sem verður aðallega á fyrsta þriðjungi meðgöngu.20 Nokkrar stúlkur með PKU eru að nálgast barnseignaaldur og þarf að sinna þessum hóp vel.
Í töflu II sést að klassísk PKU, eða alvarlegasti flokkurinn, er algengari hér en gengur og gerist meðal hvíta kynstofnsins.4 Ástæðan er trúlega sú að tvær algengustu PAH-stökkbreytingarnar á Íslandi, Y377fsdelT og P281L, valda klassískri PKU. Þessar tvær stökkbreytingar eru rúmlega 60% allra þeirra stökkbreytinga sem fundist hafa hér á landi (tafla IV). Stökkbreytingin Y377fsdelT er séríslensk og líklega komin frá Suðurlandi. Hefur genaflökt (genetic drift) leitt til þess að Y377fsdelT er jafn algeng og raun ber vitni. Ekki var um náin ættartengsl að ræða í einstaklingum fæddum fyrir 1997.1 Ef Y377fsdelT hefði ekki komið til má ímynda sér að nýgengi PKU á Íslandi væri mun lægra, þar sem 16 af 27 manns sem greinst hafa með PKU bera stökkbreytinguna. Hátt algengi P281L er líklega vegna landnemaáhrifa (founder effect). Samkvæmt setraðagreiningu virðist P281L á Íslandi ekki vera tengd sömu stökkbreytingu á Norðurlöndum.1 Í Noregi eru átta mjög algengar PAH-stökkbreytingar.21 Þrjár þeirra, R408W, Y414C, og IVS12nt1, hafa greinst á Íslandi (tafla IV) en eru einnig algengar í öðrum löndum. F299C er ein af algengustu PAH-stökkbreytingum Noregs og er þriðja algengasta stökkbreytingin hérlendis.21 Þess má geta að um 6,1% allra stökkbreytinga sem fundust á Írlandi má tengja við Skandinavíu sem gefur líklega til kynna áhrif strandhögga víkinga á írska mannfræðisögu.22 Árið 1997 sást að enga PKU-samsætu hérlendis væri hægt tengja við Írland. Sömu gerðir stökkbreytinga höfðu ekki sömu setraðirnar. Hins vegar mátti tengja margar stökkbreytingar við Noreg.1 Til þess að geta staðhæft það sama um þær 20 samsætur sem greinst hafa hér á landi eftir 1997 þyrfti að setraðagreina þær.
Meðferðarmöguleikar PKU hafa aukist undanfarin ár. Auðvelt er að fá nokkuð fjölbreytt Phe-snautt fæði. Í dag taka nokkrir Íslendingar með PKU stórar hlutlausar amínósýrur og geta þar af leiðandi gefið aðeins eftir í hefðbundinni meðferð. Við BH4-gjöf öðlast einstaklingarnir mikið frelsi sem öðrum finnst annars sjálfgefið eins og að borða úr öllum fæðuflokkum, þurfa ekki að hafa hugann við hvað borðað er öllum stundum eða taka inn mikið magn af töflum og blöndum. Einnig verða síðkomnir fylgikvillar PKU ólíklegri, sérstaklega ef BH4-meðferð er beitt fyrstu æviárin.10 Samkvæmt töflu III svöruðu tveir arfblendnir Y377fsdelT-berar ekki BH4-hleðsluprófi. Þeir bera báðir P281L á hinni samsætunni en vitað er að P281L svarar BH4-hleðsluprófi ekki.23 Niðurstaða okkar sýnir að íslenska stökkbreytingin Y377fsdelT svarar ekki BH4-meðferð. BH4-svörun Y414C var áður þekkt.12 Tveir úr þýði okkar með Y414C svara BH4-meðferð (tafla III). Fjórir til viðbótar bera stökkbreytingar sem þekktar eru af því að svara BH4-hleðsluprófi, þrír með R68S og einn með A403V.12, 23 Áhugavert væri að sjá hvort þeir svari BH4-hleðsluprófi og geti því hafið BH4-meðferð.
Ungt fólk sem glímir í dag við PKU á oft í miklum erfiðleikum með að stjórna meðferð sinni á fullnægjandi hátt þrátt fyrir hjálp foreldra og aðstandenda.24 Margir þeir sem eru með PKU á Íslandi eru á unglingsaldri eða að nálgast hann. Huga má að þessum aldurshópi enn frekar, veita frekari fræðslu og aðstoð til að ná fullum tökum á meðferðinni.
Þakkir fá þátttakendur og aðstandendur þeirra, Ásgeir Haraldsson og Jón Jóhannes Jónsson.
Heimildir
- Guldberg P, Zschocke J, Dagbjartsson A, Henriksen KF, Guttler F. A molecular survey of phenylketonuria in Iceland: identification of a founding mutation and evidence of predominant Norse settlement. Eur J Hum Genet 1997; 5: 376-81.
- Erlandsen H, Patch MG, Gamez A, Straub M, Stevens RC. Structural studies on phenylalanine hydroxylase and implications toward understanding and treating phenylketonuria. Pediatrics 2003; 112: 1557-65.
- Bowden JA, McArthur CL, 3rd. Possible biochemical model for phenylketonuria. Nature 1972; 235: 230.
- Hanley WB. Adult phenylketonuria. Am J Med 2004; 117: 590-5.
- Kayaalp E, Treacy E, Waters PJ, Byck S, Nowacki P, Scriver CR. Human phenylalanine hydroxylase mutations and hyperphenylalaninemia phenotypes: a metanalysis of genotype-phenotype correlations. Am J Hum Genet 1997; 61: 1309-17.
- Scriver CR, Sly S, eds. The Metabolic and Molecular Bases of Inherited Disease. 8.ed. McGraw-Hill Professional, New York 2000.
- Antoshechkin AG, Chentsova TV, Tatur V, Naritsin DB, Railian GP. Content of phenylalanine, tyrosine and their metabolites in CSF in phenylketonuria. J Inherit Metab Dis 1991; 14: 749-54.
- Holtzman NA, Welcher DW, Mellits ED. Termination of restricted diet in children with phenylketonuria: a randomized controlled study. N Engl J Med 1975; 293: 1121-4.
- Horner FA, Streamer CW, Clader DE, Hassell LL, Binkley EL Jr, Dumars KW Jr. Effect of phenylalanine-restricted diet in phenylketonuria. II. AMA J Dis Child 1957; 93: 615-8.
- Abadie V, Berthelot J, Feillet F, et al. [Management of phenylketonuria and hyperphenylalaninemia: the French guidelines]. Arch Pediatr 2005; 12: 594-601.
- National Institutes of Health Consensus Development Conference Statement: phenylketonuria: screening and management, October 16-18, 2000. Pediatrics 2001; 108: 972-82.
- Blau N, Erlandsen H. The metabolic and molecular bases of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Mol Genet Metab 2004; 82: 101-11.
- Bremer HJ, Marx D, Nutzenadel W, Bickel H. [Thinlayer chromatography of urinary amino acids]. Klin Wochenschr 1970; 48: 682-8.
- Bernegger C, Blau N. High frequency of tetrahydrobiopterin-responsiveness among hyperphenyla-aninemias: a study of 1,919 patients observed from 1988 to 2002. Mol Genet Metab 2002; 77: 304-13.
- O‘Neill CA, Eisensmith RC, Croke DT, Naughten ER, Cahalane SF, Woo SL. Molecular analysis of PKU in Ireland. Acta Paediatr Suppl 1994; 407: 43-4.
- Ozalp I, Coskun T, Tokatli A, et al. Newborn PKU screening in Turkey: at present and organization for future. Turk J Pediatr 2001; 43: 97-101.
- Dobson JC, Williamson ML, Azen C, Koch R. Intellectual assessment of 111 four-year-old children with phenylketonuria. Pediatrics 1977; 60: 822-7.
- Waisbren SE, Levy HL. Agoraphobia in phenylketonuria. J Inherit Metab Dis 1991; 14: 755-64.
- Thompson AJ, Smith I, Brenton D, et al. Neurological deterioration in young adults with phenylketonuria. Lancet 1990; 336: 602-5.
- Stevenson RE, Huntley CC. Congenital malformations in offspring of phenylketonuric mothers. Pediatrics 1967; 40: 33-45.
- Eiken HG, Knappskog PM, Boman H, et al. Relative frequency, heterogeneity and geographic clustering of PKU mutations in Norway. Eur J Hum Genet 1996; 4: 205-13.
- O‘Donnell KA, O‘Neill C, Tighe O, et al. The mutation spectrum of hyperphenylalaninaemia in the Republic of Ireland: the population history of the Irish revisited. Eur J Hum Genet 2002; 10: 530-8.
- Bardelli T, Donati MA, Gasperini S, et al. Two novel genetic lesions and a common BH4-responsive mutation of the PAH gene in Italian patients with hyperphenylalaninemia. Mol Genet Metab 2002; 77: 260-6.
- Weglage J, Fünders B, Ullrich K, Rupp A, Schmidt E. Psychosocial aspects in phenylketonuria. Eur J Pediatr 1996; 155 Suppl 1: S101-4.