
01. tbl. 97. árg. 2011
Wernickes encephalopathy in chronic alcoholics
Complete version in English pdf
Abstract
Wernicke's encephalopathy (WE) is caused by thiamine (vitamin B) deficiency and most commonly found in individuals with chronic alcoholism and malnutrition. Clinically, its key features are mental status disorders and oculomotor abnormalities as well as stance and gait ataxia. The diagnosis of WE is frequently missed although delay of appropriate treatment can lead to death or Korsakoff´s amnestic syndrome. It is therefore crucial in suspected cases of WE, not to await confirmation of diagnosis, but immediately administer high-dose intravenous thiamine and simultaneously treat magnesium deficiency. Alcoholics at risk of WE should immediately on admission receive prophylactic therapy with parenteral thiamine
Introduction
Wernicke‘s encephalopathy (WE) is caused by thiamine (vitamin B1) deficiency, deriving from malnutrition of various sources and most commonly found in patients with chronic alcoholism. WE is a clinically underdiagnosed, acute or subacute illness that can cause permanent memory disturbance or death if proper treatment is not given in time. Thiamine deficiency can also cause cerebellar degeneration (alcoholic cerebellar degeneration, nutritional cerebellar degeneration) and neuropathy (alcohol neuropathy, thiamine deficiency neuropathy). 1
WE was first described in 1881 by the German physician Carl Wernicke (1848-1905) as an acute illness affecting two men with chronic alcoholism and a young woman with protracted vomiting after drinking sulfuric acid in a suicide attempt. Their symptoms and signs were acute confusion, impairment of consciousness, paresis of ocular muscles, nystagmus and unsteady or ataxic gait. All three died after a short course of illness. Neuropathology revealed punctate hemorrhages around the third and fourth ventricles and aqueduct. Wernicke named the disease “acute superior hemorrhagic polioencephalitis”.2, 3 During 1887 through 1889, a Russian physician Sergei S. Korsakoff (1853-1900) described a similar illness in a larger group of patients with acute confusion and peripheral neuropathy. Those who survived had protracted memory disturbance with great difficulty in memorizing recent events. He named the disease “polyneuritic psychosis”4. In the first decades of the 20th century it became widely accepted that the acute disease named Wernicke's encephalopathy is caused by a deficiency of thiamine and that Korsakoff´s amnestic syndrome or Korsakoff´s psychosis (KS) is its chronic sequel Should symptoms of Korsakoff´s amnestic syndrome develop, the entire disease process is called Wernicke-Korsakoff syndrome.1
Epidemiology
Information on the prevalence and epidemiology of WE derives for the most part from neuropathologic studies. Lifetime prevalence of WE in the developed world is probably around 1.0% (0.4-2.8% according to various studies).1, 5-9 Most patients have a clear history of malnutrition and 77-90% have chronic alcoholism.6, 10 In neuropathologic studies, WE is found in 8.9-12.5% of individuals with chronic alcoholism5, 11. It is also seen in non-alcoholic patients with malnutrition of various causes, such as protracted starvation12 or hyperemesis gravidarium13, 14. WE can be a consequence of gastrointestinal operations for morbid obesity or other operations on the gastrointestinal tract.15 It can also result from infusion of glucose solutions16 or parenteral nutrition 17 without proper thiamine supplementation. It has been diagnosed among patients on hemo- and peritoneal dialysis18, 19 in patients with various cancers, after bone marrow transplantation 20-22 and in patients with AIDS.23, 24
Pathophysiology
Thiamine is a water soluble vitamin, absorbed in the small intestine, and is found in various food products. The daily requirements for adults is approximately 1 mg and total body stores are 25-30 mg. Severe deficiency of thiamine develops in three weeks if ingestion shuts down 25. Thiaminediphosphate (TDP) is the active form of intracellular thiamine. It is a vital cofactor of transketolase (TK), pyruvate dehydrogenase (PDH) and a-Ketoglutarate dehydrogenase (α-KGDH) that are key enzymes in glucose- and amino acid metabolism. Thiamine requirements increase with increased metabolic rate, e.g. in systemic infection.25
Chronic alcoholics have a lower ratio of thiamine in its active form of TDP and also a lower fractional increase of TDP after thiamine injections. Alcoholics with thiamine deficiency require therefore a higher dose of thiamine to respond similarly to normal controls 26 Furthermore, magnesium is a necessary cofactor in the formation of TDP and magnesium deficiency, common among alcoholics, appears to increase the neurological damage in thiamine deficiency.27-30 Response to thiamine treatment can be inhibited by concurrent magnesium deficiency.31-33 It may therefore be important to correct magnesium deficiency in addition to thiamine therapy in WE patients.
There are several reasons for the higher rate of WE in patients with chronic alcoholism. Nutritional deficiency with coexisting decrease in thiamine intake, vomiting or diarrhea are common in chronic alcoholism.34 In malnourished individuals the ability of the gastrointestinal tract to absorb a fixed dose of thiamine is diminished and does not correct fully until after 6-8 weeks of a healthy nutritional diet.35-37 The liver stores a large part of the body's supplies of thiamine and if diseased its capacity to store and handle thiamine diminishes.34, 37, 38 Alcohol withdrawal, delirium tremens and infections all increase metabolic rate and requirements for thiamine 1, 25. The formation of active thiamine requiring enzymes (TK, PDH and α-KGDH) is inhibited in alcoholics, due to the reduced utilization of thiamine (lower fraction of thiamine in the active form of TDP and magnesium deficiency ). Chronic alcoholism causes up-regulation of NMDA-receptors that appears to increase the neurological damage in thiamine deficiency, through NMDA-receptor mediated excitotoxicity. 28, 29, 39
Pathologic features
Histological changes in WE are of two main types: acute and chronic or permanent.
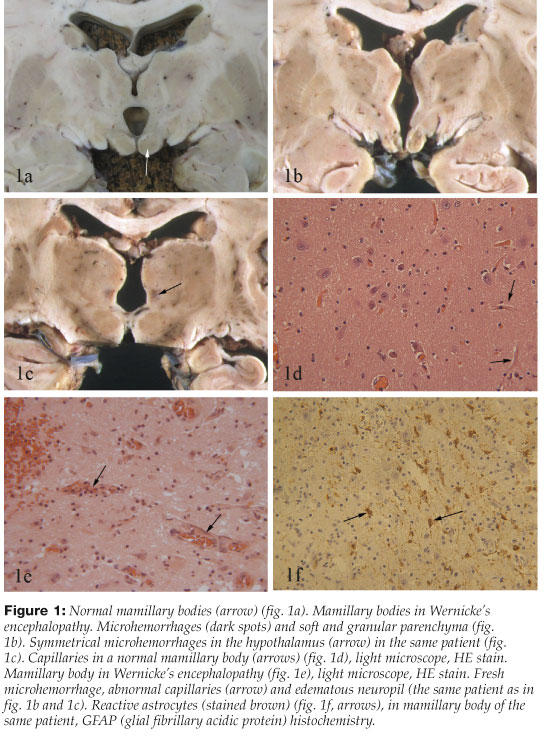
Acute changes are most frequently seen in the mamillary bodies, around the third and fourth ventricles and aqueduct of the midbrain. Specific changes in these areas are diagnostic of WE1,,10. Punctate hemorrhages in the mamillary bodies (10%) or around the third ventricle are visible to the naked eye, but in 30% of cases the brain, including the mamillary bodies, has a normal macroscopic appearance1, 10 (Figure 1a-1c). Microscopic examination is necessary for proper diagnosis and shows, almost without exception, symmetric histopathological changes in the mamillary bodies and most often in other areas as well, including the hypothalamus, thalamus (typically dorsomedial nuclei), around the aqueduct of the midbrain, in the oculomotor nuclei, nuclei dorsalis of the vagus, in the vestibular nuclei and the nuclei of the abducens nerves.1, 10, 40 On microscopic examination acute vascular and neuropil changes predominate. Capillaries are pathologically prominent due to endothelial hypertrophy and possibly capillary proliferation. Edema, extravasation of red cells and microhemorrhages around capillaries are also seen. Astrocytic reaction soon becomes prominent with destruction of myelin and axons, but seldom reduction of nerve cells in the mamillary bodies 1-3, 7, 10, 41 (Figure 1d-1f).
{kind=link}
Permanent (chronic) changes are characterized by destruction of nerve tissue and gliosis in the areas mentioned above. Atrophy of the mamillary bodies is visible to the naked eye as well as widening of the third and fourth ventricles and the aqueduct of the midbrain. On microscopic examination there is proliferation of astrocytes, tissue destruction and gliosis. (Figure 1f). In the chronic histopathological changes of WE, the capillary endothelium appears normal and there are no pathologic fresh pericapillary hemorrhages. 1, 10, 40, 41 Chronic changes are often confined to the mamillary bodies and the dorsomedial nuclei of the thalami, opposed to acute changes that are typically found more widely, including the brain stem.42
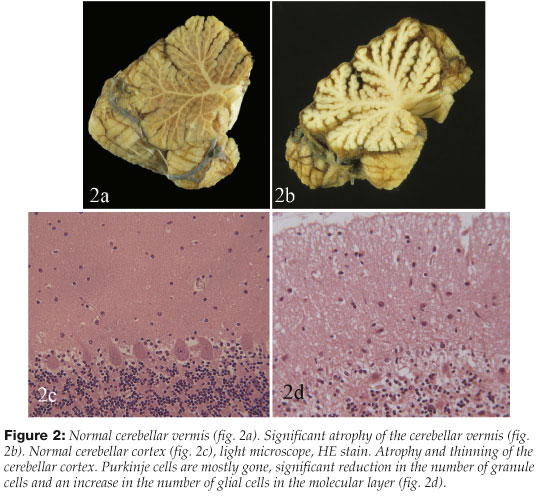
Autopsy on individuals with the neuropathologic changes of WE often shows degeneration of the anterior-superior part of the cerebellar vermis. It is visible to the naked eye in slightly more than one third and in almost half on microscopic examination1, 10 (Figure 2).
{kind=link}
Clinical features
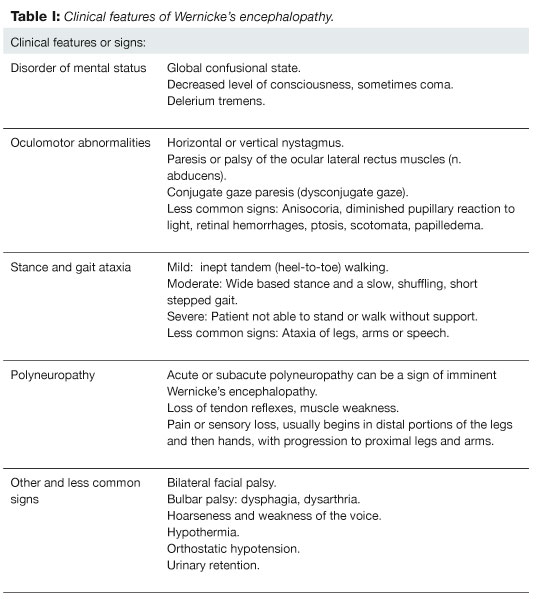
The clinical features of WE present acutely or subacutely (Table I). Its cardinal features are: 1) disorder of mental status or confusion, 2) oculomotor abnormalities and 3) stance and gait ataxia. Usually on examination only one or two of these cardinal features of WE are found and all three only in one third of the patients.1
{kind=link}
Disorder of mental status: Most common is a global confusional state with lethargy and apathy, often without prominent agitation. Attention, concentration and memory are disturbed, and in more severe cases delerium tremens, stupor or coma are present.1
Oculomotor abnormality: Horizontal gaze evoked nystagmus is most common and many have vertical nystagmus as well. Also frequent are paresis or palsy of the ocular lateral rectus muscles (abducens nerves) and conjugate gaze paresis (dysconjugate gaze). Other less common ocular signs include: anisocoria, diminished pupillary reaction to light, retinal hemorrhages, ptosis, scotomata and papilledema.1 In severe illness complete ocular palsy, absent vestibulo-ocular response (absent doll's sign or absent response to ice-water caloric testing of vestibulo-ocular function) or hypothermia and hypotension can be present.43
Stance and gait ataxia of variable degree is seen in the majority of patients. Its severity varies from mild imbalance and unsteadiness with difficulty in tandem (heel-to-toe) walking to wide-based shuffling or ataxic gait. In the most severe cases the patient is unable to sit up in bed, to stand or walk, even with considerable assistance.1
In acute WE ataxia can be present in the extremities, most often seen on heel-knee to shin test but ataxic speech (scanning or slurred speech) is rare 1. The imbalance when sitting, standing or walking is believed to be due to an acute dysfunction in the vestibular nuclei. Two studies have shown that a vestibular nuclear dysfunction is almost a universal finding in patients with acute WE. It can be confirmed by ice-water caloric testing, or similar tests, of vestibulo-ocular function. In WE there is a reduced or absent vestibulo-ocular response on ice-water irrigation of the ear-canal. After thiamine treatment and recovery from the acute phase the vestibule-ocular response can take up to two months to normalize.44, 45 On the other hand, the chronic gait disturbance often present after recovery from acute WE (and commonly observed in chronic alcoholics without prior WE) is believed to be caused by degeneration of the cerebellar vermis (not by vestibular dysfunction).1
Polyneuropathy is seen among 60-82% of patients with WS, either affecting only the legs (57%) or both arms and legs (25%).1, 46 Acute or subacute polyneuropathy can be seen early in or shortly before the presentation of WE 4, 47. Korsakoff originally described polyneuropathy as a presenting feature, hence the name “psychosis polyneuritica”.4 The neuropathy is characterized by reduced or absent heel tendon reflexes, reduced sensation or pain in the distal feet and sometimes muscle weakness. Sensory disturbances begin in the feet and can progress proximally, affecting fingers and hands and with continued progression the proximal arms and legs.
Autonomic neuropathy is less common in WE. It can appear in the sympathetic system with orthostatic hypotension or the parasympathetic with urinary retention. Affection of the vagus nerve can cause dysphagia, hoarseness or weakness of the voice.48
Other less common signs: Hypothermia is rare (1-4%), believed to be due to a lesion in the posterior part of the hypothalamus.1, 10, 49 Hypotension is seen among 2% of patients. Bilateral facial paresis, bulbar paresis 1 or extremity paresis with increased tendon reflexes and positive Babinski 50 are also rare.
Sequel of Wernicke's encephalopathy
If an individual with an acute WE does not receive thiamine through food or treatment, he will die. Even with treatment, permanent damage is common 1. Korsakoff´s amnestic syndrome (KS) is a well known sequel of acute WE, characterized by difficulty in remembering events (episodic memory) prior to (retrograde amnesia) and following (anterograde amnesia) the acute illness. Patients with KS have diminished episodic memory and reduced ability to memorize or learn new things. They have little insight into and are indifferent to their disability and their initiative and spontaneity may be reduced to the point of apathy.1, 4. In the most comprehensive study of the acute treatment and sequel of WE to date, patients were treated with a much smaller dose of thiamine (50-100 mg daily), than is currently recommended (600-1500 mg daily). In this study the patients´ prospects were dismal, 24% died and 81% of the survivors developed KS.1 The severity of their amnestic syndrome was varied and so were the degrees of memory function recovery during the first months after the illness. Recovery of memory function among patient with KS ranged from complete (21%), to significant (25%), slight (28%) or none (26%)1. The majority were left with a chronic gait disturbance (62%), characterized by a slow, wide-based gait and abnormal performance on tandem walking.1 Ocular paresis resolved soon after treatment with thiamine, recovery often starting within a few hours. Nystagmus improved more slowly and in 60% it was permanent.1
Diagnosis
The diagnosis of acute WE is frequently missed by physicians. One study showed that only 20% of cases were correctly diagnosed before death, although the great majority was evaluated by a physician in hospital shortly before dying 51. In clinical studies only one third of the cases have all three main features of the disease 1 and in retrospective studies of pathologically diagnosed cases only 16% have all three main clinical features of WE noted in their charts.51
There is a possible lack of proper neurological evaluation of alcoholics with WE, as disturbances of mental status are the most often recorded clinical features in the charts but features requiring more thorough neurologic evaluation like nystagmus, ataxia and gait disturbance are more seldom noted.5, 6, 11, 51
It is important for physicians to recognize the clinical features of WE and consider this diagnosis when chronic alcoholics present with confusion or gait disturbance. They should do a thorough neurological evaluation in the attempt to reveal the more specific signs of WE and keep in mind that only a minority of patients with WE have all the main clinical features of the disease at presentation. It is important to remember that some of the typical features of WE can easily be attributed to intoxication with alcohol or drugs, infection, electrolyte imbalance, etc.
Neurologic evaluation: Changes in mental status, such as decreased level of consciousness and disorientation should be noted. Particular attention should be paid to a presence of ocular muscle paresis or nystagmus and the patient's ability to sit up in bed, stand and walk normally and unaided, including tandem walking. After clinical evaluation ancillary tests should be ordered according to likely differential diagnoses and possible complications of WE. Serum concentration of magnesium should be ordered and there should be a low threshold for an acute CT brain scan. MRI of the brain with contrast should be ordered acutely or subacutely.
A presumptive diagnosis of WE: In 2001 The Royal College of Physicians published guidelines on the diagnosis and treatment of WE in accident and emergency departments.52 Important additions to those are recommendations by Thomson and Marshall on the treatment of patients at risk of developing Wernicke's encephalopathy in the community.53 Accordingly, only a presumptive diagnosis of WE (a presumed WE) is needed to treat patients immediately with high dose thiamine. Patients having a history or signs of chronic alcohol misuse and any of the following unexplained symptoms: Acute confusion (not due to intoxication), delirium tremens, memory disturbance, decreased conscious level, opthalmoplegia or nystagmus, ataxia (not due to intoxication) or unexplained hypothermia with hypotension should be presumed to have WE and treated accordingly. (Table II).52, 53 It should be noted that the ataxia in WE is characterized by imbalance or ataxia on sitting up in bed or standing and walking and is most likely due to acute bilateral vestibular paresis, often with little or no ataxia in the extremities on heel-knee to shin testing or finger-nose testing.1
{kind=link}
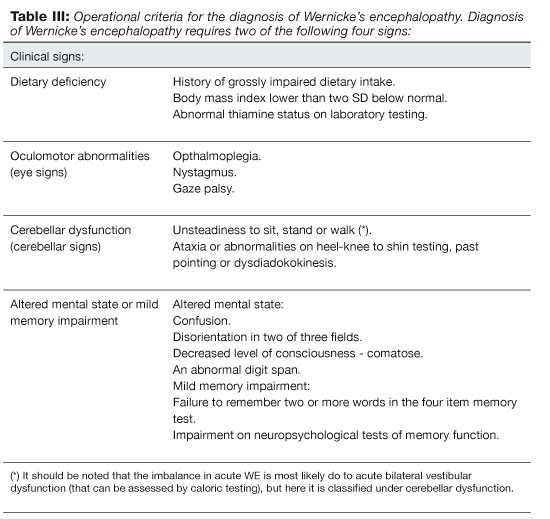
Caine et al. published operational criteria for the diagnosis of WE and KS (Wernicke-Korsakoff syndrome) among alcoholics in 1997 (Table III). Their purpose was to increase the precision of clinical diagnosis. According to them two out of the four following have to be present for diagnosis: 1) Dietary deficiency, 2) oculomotor abnormalities (eye signs), 3) cerebellar dysfunction (cerebellar signs) or 4) altered mental state or mild memory impairment. KS is diagnosed if the patient fulfills the criteria for WE and additionally has memory impairment and disorientation without being in a confusional state. In KS the patient has a normal level of consciousness and memory impairment does not resolve with thiamine treatment. It is noteworthy that the diagnostic criteria were validated retrospectively on 106 deceased alcoholic patients by reviewing charts and comparing them to neuropathologic results. The sensitivity of the criteria was 85% and specificity 100% in diagnosing WE and the sensitivity was 88% for diagnosing KS. Notably, the sensitivity was only 50% in diagnosing WE if there was a concurrent hepatic encephalopathy. The similarity of clinical features in patients with hepatic encephalopathy and WE is a likely explanation for the low sensitivity.54 These diagnostic criteria have not been validated prospectively and they probably do not have high specificity in diagnosing acute WE from other causes of encephalopathy in alcoholics, particularly in alcoholics with chronic sequel, like nystagmus or gait disturbance, after prior episodes of WE or alcoholic cerebellar degeneration. The criteria's main strength, however, is ensuring that most patients with acute WE receive proper therapy and are therefore less likely to end up with disabling neurological sequel. Many patients will be given unnecessarily high doses of thiamine, but thiamine therapy is not costly and seldom has serious adverse effects.
{kind=link}
Laboratory measurement of thiamine deficiency: Thiamine deficiency can be assessed by measuring the activity of transketolase (TK) in hemolysed blood. In thiamine deficiency the activity of TK is significantly diminished in the hemolysate and shows abnormal increase in enzyme activity (>25%) with the addition of a saturating dose of TDP (increased TDP effect). 55, 56 The concentration of thiamine, thiamine monophosphate and TDP can also be measured directly in blood or hemolysed erythrocytes with chromatography.57 These methods have limited or no role in the clinical diagnosis of acute WE.
Magnetic resonance imaging (MRI): The diagnosis of WE is clinical, based on history, neurologic evaluation, response to thiamine treatment and characteristic evolution of the illness. MRI is of major importance for the diagnosis, especially if neurological expertise or neurological evaluation is limited or the disease presentation atypical. MRI may also reveal other possible causes of the symptoms.
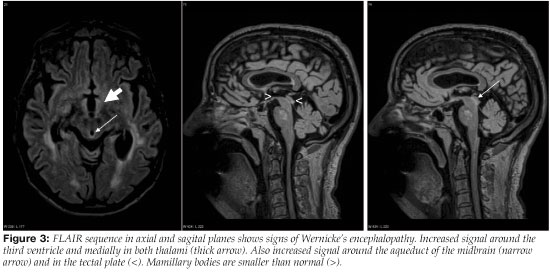
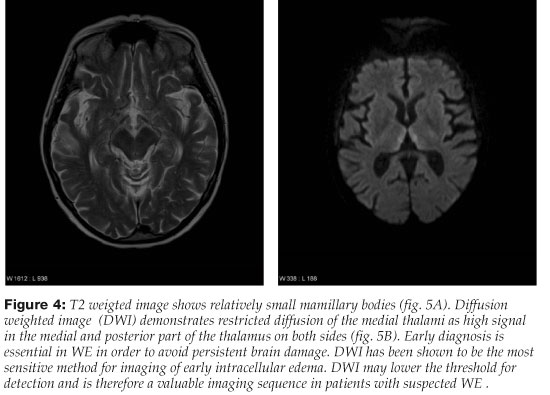
The typical MRI changes in acute WE are symmetric signal changes (signal increasement on T2 weighted and decreasement on T1 weighted sequences) around the third ventricle, in the mamillary bodies, medial thalami and around the aqueduct of the midbrain (areas of neuropathological lesions).50, 58, 59 Changes in these areas can also be discerned on FLAIR sequences or diffusion-weighted imaging59, 60 (Figures 3 and 4). Contrast enhancement is seen among 63-67% of patients that have MRI changes in acute WE. It is most often limited to the mamillary bodies and is sometimes the only diagnostic change seen on MRI.50, 59 In two studies on the role of MRI in the diagnosis of WE, sensitivity of MRI was 53-58% and specificity 93% in revealing changes corresponding to WE.61, 62 Therefore, patients with acute WE commonly have a normal MRI which does not rule out a possible diagnosis of an acute WE.
{kind=link}
{kind=link}
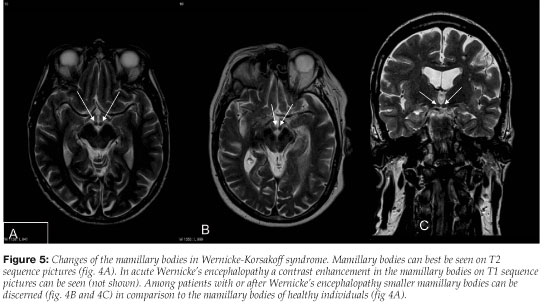
In the course of the illness (weeks to months), an MRI often shows degenerative changes in the brain (atrophy of the mamillary bodies, enlargement of the third ventricle or aqueduct and possibly degeneration of the cerebellar vermis) 63 (Figure 5). These changes, when seen on MRI during the illness can strongly support or confirm the clinical diagnosis of WE .64-68
{kind=link}
Confirmation of a presumed WE: It should be noted that response to thiamine, how the disease resolves, and possible neurological sequel are highly characteristic of WE and can be used in addition to possible changes on MRI or neuropathology to confirm the clinical diagnosis of a presumed WE (Table IV).
{kind=link}
Differential diagnosis
Intoxication with alcohol or drugs can cause all the main features of WE. Some diseases can also be clinically similar to WE or KS, or may show similar changes on MRI, the most important being 1) Bilateral paramedian thalamic infarcts that can have similar clinical features as an acute WE 69-71, 2) anterior thalamic or mamillothalamic tract infarcts that can give similar clinical features as KS 69, 72, 73, 3) cytomegalovirus (CMV) encephalitis, well known in immunocompromised patients, especially in AIDS, can both have similar clinical features and show similar changes on MRI as an acute WE 74, 75, 4) demyelinating diseases, neurological Behçet's disease and herpes (HSV) encephalitis are also possible differential diagnoses, 5) Hepatic encephalopathy can be difficult to distinguish clinically from an acute WE 54, especially among alcoholics that have had a prior episode or history of WE or alcoholic cerebellar degeneration, with chronic sequel of nystagmus or stance and gait ataxia.
Treatment of likely Wernicke's encephalopathy
All alcoholics with a likely WE should be treated with high doses of thiamine without delay. Included are alcoholics with a presumed acute WE (Table II), serious head injury or other conditions that make proper neurologic evaluation impossible, resulting in the inability to exclude the diagnosis of WE with reasonable confidence.52 Physicians should also be alert to the possibility of acute WE in alcoholics with hepatic encephalopathy, even treat them with high doses of thiamine and assess their clinical response.54 Thiamine should never be given by mouth in acute WE as the absorption of thiamine from the gastrointestinal tract in alcoholics is low and unpredictable. Thiamine should only be given intravenously or intramuscularly.36, 52, 76
Recommended thiamine treatment: There are no randomized controlled clinical trials or other reliable clinical studies on the critical dose of thiamine needed to minimize disabling neurological outcome after acute WE.77 Recent guidelines concerning the proper dose of thiamine in WE are founded on several factors, all of which support the use of much higher doses than have previously been given: 1) The knowledge of the dismal prospects for patients if treated with the lower doses (50-100 mg daily) previously used, 2) better knowledge of the pharmacodynamics of thiamine in thiamine deficient patients, 3) the differences in the pharmaodynamics of thiamine in alcoholics compared to non-alcoholics, 4) published cases and clinical experience (including our own experience of unpublished Icelandic cases) that point toward reduced neurological sequel following higher doses of thiamine and 5) low frequency of adverse effects with parenteral thiamine therapy. It should be noted that anaphylaxis is a very rare adverse effect when thiamine is given intravenously or intramuscularly.35, 39, 52, 78-81
Two guidelines have recently been published on treatment for acute WE. One recommends thiamine hydrochloride 500 mg given three times daily intravenously for three consecutive days. If the response is positive, the dose should then be lowered to 500 mg daily given intravenously or intramuscularly for five more days. If there is no response after the first three days, the treatment should be stopped, and an alternative diagnosis sought.52 The other recommends thiamine hydrochloride 200 mg given three times daily intravenously until there is no further improvement in signs and symptoms.82 Thiamine hydrochloride can be administrated slowly intravenously80, 81 but both guidelines recommend infusion of thiamine diluted with normal saline or 5% glucose given over 30 minutes.
Correction of magnesium deficiency with intravenous infusion of magnesium sulfate seems logical in acute WE as magnesium deficiency appears to increase the neurological damage in thiamine deficiency 27-30 and the response to thiamine treatment can be deficient in patients with significant magnesium deficiency.31-33. Additional studies on the role of magnesium deficiency in WE and the neurological damage in chronic alcoholism seems a promising field for further studies. Correction of magnesium deficiency with oral therapy to minimize the damage in WE is inappropriate because of the high probability of absorption disturbance and the need for a rapid correction in acute WE. It takes several days of parenteral therapy to correct the intracellular magnesium deficiency.83, 84
Prophylactic therapy
Prophylactic thiamine should be given immediately on admission to all alcoholics with a significant risk of WE, including those needing medical detoxification, having evidence of nutritional deficiency or receiving intravenous glucose solution. Loss of appetite, nausea and vomiting can be the first signs of thiamine deficiency.85 As discussed previously it is inappropriate to give admitted alcoholics oral thiamine to prevent WE. Thiamine hydrochloride 200-250 mg should be given daily for three to five days intramuscularly to minimize the risk of WE in admitted alcoholics.53, 79, 86
It is reasonable to recommend patients with active chronic alcoholism to take vitamin supplements containing at least 15 mg of thiamine daily. Thiamine supplementation can reduce the risk of their developing thiamine deficiency which causes many of the chronic disabling neurological complications of alcoholism.1, 26 To increase compliance it is advisable to ask close relatives to buy the supplements or include them with other medicines in a medical dosing from a pharmacy.
Authors would like to thank Jónas Knútsson and Nick Cariglia for assistance with translation.
References:
- Victor M, Adams R, Collins G. The Wernicke-Korsakoff 1. Victor M, Adams R, Collins G. The Wernicke-Korsakoff syndrome and related disorders due to alcoholism and malnutrition. 2 ed. F.A. Davis Company, Fíladelfíu 1989.
- Wernicke C. Die acute, hämorrhagische Polioencephalitis superior. In: Lehrbuch der Gehirnkrankheiten fur Aerzte und Studirende, vol 2. Theodor Fischer, Berlin, Kassel 1881: 229-42.
- Thomson AD, Cook CCH, Guerrini I, Sheedy D, Harper C, Marshall EJ. Wernicke‘s encephalopathy revisited. Translation of the case history section of the original manuscript by Carl Wernicke ‚Lehrbuch der Gehirnkrankheiten fur Aerzte and Studirende‘ (1881) with a commentary. Alcohol Alcohol 2008; 43: 174-9.
- Victor M, Yakovlev PI. S.S. Korsakoff‘s psychic disorder in conjunction with peripheral neuritis; a translation of Korsakoff‘s original article with comments on the author and his contribution to clinical medicine. Neurology 1955; 5: 394-406.
- Torvik A, Lindboe CF, Rogde S. Brain lesions in alcoholics. A neuropathological study with clinical correlations. J Neurol Sci 1982; 56: 233-48.
- Lindboe CF, Loberg EM. Wernicke‘s encephalopathy in non-alcoholics. An autopsy study. J Neurol Sci 1989; 90: 125-9.
- Cravioto H, Korein J, Silberman J. Wernicke‘s encephalopathy. A clinical and pathological study of 28 autopsied cases. Arch Neurol 1961; 4: 510-9.
- Victor M, Laureno R. Neurologic complications of alcohol abuse: Epidemiologic aspects. In: Schoenberg BS, ed. Advances in Neurology, Vol 19. Raven Press, New York 1978: 603-17.
- Harper C, Fornes P, Duyckaerts C, Lecomte D, Hauw JJ. An international perspective on the prevalence of the Wernicke-Korsakoff syndrome. Metab Brain Dis 1995; 10: 17-24.
- Harper C. The incidence of Wernicke‘s encephalopathy in Australia--a neuropathological study of 131 cases. J Neurol, Neurosurg Psychiatr 1983; 46: 593-8.
- Lindboe CF, Loberg EM. The frequency of brain lesions in alcoholics. Comparison between the 5-year periods 1975-1979 and 1983-1987. J Neurol Sci 1988; 88: 107-13.
- Doraiswamy PM, Massey EW, Enright K, et al. Wernicke-Korsakoff syndrome caused by psychogenic food refusal: MR findings. Am J Neuroradiol 1994; 15: 594-6.
- Selitsky T, Chandra P, Schiavello HJ. Wernicke‘s encephalopathy with hyperemesis and ketoacidosis. Obstet Gynecol 2006; 107: 486-90.
- Chiossi G, Neri I, Cavazzuti M, Basso G, Facchinetti F. Hyperemesis gravidarum complicated by Wernicke encephalopathy: background, case report, and review of the literature. Obstet Gynecol Surv 2006; 61: 255-68.
- Singh S, Kumar A. Wernicke encephalopathy after obesity surgery: a systematic review. Neurology 2007; 68: 807-11.
- Koguchi K, Nakatsuji Y, Abe K, Sakoda S. Wernicke‘s encephalopathy after glucose infusion. Neurology 2004; 62: 512.
- Francini-Pesenti F, Brocadello F, Famengo S, Nardi M, Caregaro L. Wernicke‘s encephalopathy during parenteral nutrition. JPEN J Parenter Enteral Nutr 2007; 31: 69-71.
- Jagadha V, Deck JH, Halliday WC, Smyth HS. Wernicke‘s encephalopathy in patients on peritoneal dialysis or hemodialysis. Ann Neurol 1987; 21: 78-84.
- Barbara PG, Manuel B, Elisabetta M, et al. The suddenly speechless florist on chronic dialysis: the unexpected threats of a flower shop? Diagnosis: dialysis related Wernicke encephalopathy. Nephrol Dial Transplant 2006; 21: 223-5.
- Boniol S, Boyd M, Koreth R, Burton GV. Wernicke encephalopathy complicating lymphoma therapy: case report and literature review. South Med J 2007; 100: 717-9.
- Onishi H, Kawanishi C, Onose M, et al. Successful treatment of Wernicke encephalopathy in terminally ill cancer patients: report of 3 cases and review of the literature. Support Care Cancer 2004; 12: 604-8.
- Bleggi-Torres LF, de Medeiros BC, Werner B, et al. Neuropathological findings after bone marrow transplantation: an autopsy study of 180 cases. Bone Marrow Transplant 2000; 25: 301-7.
- Alcaide ML, Jayaweera D, Espinoza L, Kolber M. Wernicke's encephalopathy in AIDS: a preventable cause of fatal neurological deficit. Int J STD AIDS 2003; 14: 712-3.
- Butterworth RF, Gaudreau C, Vincelette J, Bourgault AM, Lamothe F, Nutini AM. Thiamine deficiency and Wernicke's encephalopathy in AIDS. Metab Brain Dis 1991; 6: 207-12.
- Butterworth RF. Thiamin. In: Shils ME, M S, C RA, B C, J CR, eds. Modern Nutrition in Health and Disease: Lippincott Williams & Wilkins; 2005: 426-33.
- Tallaksen CM, Bohmer T, Bell H. Blood and serum thiamin and thiamin phosphate esters concentrations in patients with alcohol dependence syndrome before and after thiamin treatment. Alcohol Clin Exp Res 1992; 16: 320-5.
- Voskoboyev AI, Ostrovsky YM. Thiamin pyrophosphokinase: structure, properties, and role in thiamin metabolism. Ann N Y Acad Sci 1982; 378: 161-76.
- Singleton CK, Martin PR. Molecular mechanisms of thiamine utilization. Curr Mol Med 2001; 1: 197-207.
- Dodd PR, Beckmann AM, Davidson MS, Wilce PA. Glutamate-mediated transmission, alcohol, and alcoholism. Neurochem Int 2000; 37: 509-33.
- Flink EB. Magnesium deficiency in alcoholism. Alcohol Clin Exp Res 1986; 10: 590-4.
- Zieve L. Influence of magnesium deficiency on the utilization of thiamine. Ann N Y Acad Sci 1969; 162: 732-43.
- Traviesa DC. Magnesium deficiency: a possible cause of thiamine refractoriness in Wernicke-Korsakoff encephalopathy. J Neurol Neurosurg Psychiatry 1974; 37: 959-62.
- Dyckner T, Ek B, Nyhlin H, Wester PO. Aggravation of thiamine deficiency by magnesium depletion. A case report. Acta Med Scand 1985; 218: 129-31.
- Morgan MY. Alcohol and nutrition. Brit Med Bull 1982; 38: 21-9.
- Thomson AD. Mechanisms of vitamin deficiency in chronic alcohol misusers and the development of the Wernicke-Korsakoff syndrome. Alcohol Alcohol 2000; 35 Supplement: 2-7.
- Thomson AD, Baker H, Leevy CM. Patterns of 35S-thiamine hydrochloride absorption in the malnourished alcoholic patient. J Labor Clin Med 1970; 76: 34-45.
- Hoyumpa AM, Jr. Mechanisms of thiamin deficiency in chronic alcoholism. Am J Clin Nutr 1980; 33: 2750-61.
- Thomson AD, Pratt O. Interaction of nutrients alcohol: Absorption, transport, utilisation and metabolism. In: Watson RR, Waltz B, eds. Nutrition and alcohol.CRC Press, Boca Raton, Fl USA 1992: 75-99.
- Thomson AD, Marshall EJ. The natural history and pathophysiology of Wernicke‘s Encephalopathy and Korsakoff‘s Psychosis. Alcohol Alcohol 2006; 41: 151-8.
- Malamud N, Skillicorn SA. Relationship between the Wernicke and the Korsakoff syndrome; a clinicopathologic study of seventy cases. Arch Neurol Psychiatry 1956; 76: 585-96.
- Torvik A. Two types of brain lesions in Wernicke‘s encephalopathy. Neuropathol Appl Neurobiol 1985; 11: 179-90.
- Torvik A. Topographic distribution and severity of brain lesions in Wernicke‘s encephalopathy. Clin Neuropathol 1987; 6: 25-9.
- Wallis WE, Willoughby E, Baker P. Coma in the Wernicke-Korsakoff syndrome. Lancet 1978; 2: 400-1.
- Ghez C. Vestibular paresis: a clinical feature of Wernicke‘s disease. J Neurol Neurosurg Psychiatry 1969; 32: 134-9.
- Goor C, Endtz LJ, Muller-Kobold MJ. Electro-nystagmography for the diagnosis of vestibular dysfunction in the Wernicke-Korsakow syndrome. Clin Neurol Neurosurg 1975; 78: 112-7.
- Groen RH, Hoff HC. Wernicke‘s disease. A catamnestic study of 50 patients. Eur Neurol 1977; 15: 109-15.
- Barry H. Wernicke‘s encephalopathy in surgical practice. Lancet 1947; 2: 278-9.
- Novak DJ, Victor M. The vagus and sympathetic nerves in alcoholic polyneuropathy. Arch Neurol 1974; 30: 273-84.
- Hunter JM. Hypothermia and Wernicke‘s encephalopathy. BMJ 1976; 2: 563-4.
- Mascalchi M, Simonelli P, Tessa C, et al. Do acute lesions of Wernicke's encephalopathy show contrast enhancement? Report of three cases and review of the literature. Neuroradiol 1999; 41: 249-54.
- Harper CG, Giles M, Finlay-Jones R. Clinical signs in the Wernicke-Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry 1986; 49: 341-5.
- Thomson AD, Cook CCH, Touquet R, Henry JA; Royal College of Physicians, London. The Royal College of Physicians report on alcohol: guidelines for managing Wernicke's encephalopathy in the accident and Emergency Department. Alcohol Alcohol 2002; 37: 513-21.
- Thomson AD, Marshall EJ. The treatment of patients at risk of developing Wernicke's encephalopathy in the community. Alcohol Alcohol 2006; 41: 159-67.
- Caine D, Halliday GM, Kril JJ, Harper CG. Operational criteria for the classification of chronic alcoholics: identification of Wernicke's encephalopathy. J Neurol Neurosurg Psychiatry 1997; 62: 51-60.
- Dreyfus PM. Clinical application of blood transketolase determinations. N Engl J Med 1962; 267: 596-8.
- Sauberlich HE. Newer laboratory methods for assessing nutriture of selected B-complex vitamins. Ann Rev Nutr 1984; 4: 377-407.
- Mancinelli R, Ceccanti M, Guiducci MS, et al. Simultaneous liquid chromatographic assessment of thiamine, thiamine monophosphate and thiamine diphosphate in human erythrocytes: a study on alcoholics. J Chromatogr B Analyt Technol Biomed Life Sci 2003; 789: 355-63.
- Gallucci M, Bozzao A, Splendiani A, Masciocchi C, Passariello R. Wernicke encephalopathy: MR findings in five patients. Am J Roentgenol 1990; 155: 1309-14.
- Zuccoli G, Santa Cruz D, Bertolini M, et al. MR Imaging Findings in 56 Patients with Wernicke Encephalopathy: Nonalcoholics May Differ from Alcoholics. Am J Neuroradiol 2009; 30: 171-6.
- White ML, Zhang Y, Andrew LG, Hadley WL. MR imaging with diffusion-weighted imaging in acute and chronic Wernicke encephalopathy. Am J Neuroradiol 2005; 26: 2306-10.
- Antunez E, Estruch R, Cardenal C, et al. Usefulness of CT and MR imaging in the diagnosis of acute Wernicke's encephalopathy. Am J Roentgenol 1998; 171: 1131-7.
- Weidauer S, Nichtweiss M, Lanfermann H, Zanella FE. Wernicke encephalopathy: MR findings and clinical presentation. Eur Radiol 2003; 13: 1001-9.
- Park SH, Kim M, Na DL, Jeon BS. Magnetic resonance reflects the pathological evolution of Wernicke encephalopathy. J Neuroimaging 2001; 11: 406-11.
- Sheedy D, Lara A, Garrick T, Harper C. Size of mamillary bodies in health and disease: useful measurements in neuroradiological diagnosis of Wernicke‘s encephalopathy. Alcohol Clin Exp Res 1999; 23: 1624-8.
- Charness ME, DeLaPaz RL. Mamillary body atrophy in Wernicke‘s encephalopathy: antemortem identification using magnetic resonance imaging. Ann Neurol 1987; 22: 595-600.
- Sullivan EV, Lane B, Deshmukh A, et al. In vivo mammillary body volume deficits in amnesic and nonamnesic alcoholics. Alcohol Clin Exp Res 1999; 23: 1629-36.
- Charness ME, DeLaPaz RL. Periodic alternating nystagmus in an alcoholic with small mamillary bodies. Neurology 1988; 38: 421.
- Charness ME. Intracranial voyeurism: revealing the mammillary bodies in alcoholism. Alcohol Clin Exp Res 1999; 23: 1941-4.
- Bogousslavsky J, Regli F, Uske A. Thalamic infarcts: clinical syndromes, etiology, and prognosis. Neurology 1988; 38: 837-48.
- Gentilini M, De Renzi E, Crisi G. Bilateral paramedian thalamic artery infarcts: report of eight cases. J Neurol Neurosurg Psychiatry 1987; 50: 900-9.
- Chung SP, Kim SW, Yoo IS, Lim YS, Lee G. Magnetic resonance imaging as a diagnostic adjunct to Wernicke encephalopathy in the ED. Am J Emerg Med 2003; 21: 497-502.
- Ghika-Schmid F, Bogousslavsky J. The acute behavioral syndrome of anterior thalamic infarction: a prospective study of 12 cases. Ann Neurol 2000; 48: 220-7.
- Yoneoka Y, Takeda N, Inoue A, et al. Acute Korsakoff syndrome following mammillothalamic tract infarction. Am J Neuroradiol 2004; 25: 964-8.
- Brechtelsbauer DL, Urbach H, Sommer T, Blümcke I, Woitas R, Solymosi L. Cytomegalovirus encephalitis and primary cerebral lymphoma mimicking Wernicke's encephalopathy. Neuroradiology 1997; 39: 19-22.
- Torgovnick J, Arsura EL, Lala D. Cytomegalovirus ventriculoencephalitis presenting as a Wernicke's encephalopathy-like syndrome. Neurology 2000; 55: 1910-3.
- Tallaksen CM, Sande A, Bohmer T, Bell H, Karlsen J. Kinetics of thiamin and thiamin phosphate esters in human blood, plasma and urine after 50 mg intravenously or orally. Eur J Clin Pharmacol 1993; 44: 73-8.
- Day E, Bentham P, Callaghan R, Kuruvilla T, George S. Thiamine for Wernicke-Korsakoff Syndrome in people at risk from alcohol abuse. Cochrane Database Syst Rev 2004: CD004033.
- Cook CC, Hallwood PM, Thomson AD. B Vitamin deficiency and neuropsychiatric syndromes in alcohol misuse. Alcohol Alcohol 1998; 33: 317-36.
- Cook CC. Prevention and treatment of Wernicke-Korsakoff syndrome. Alcohol Alcohol 2000; Suppl 35: 19-20.
- Wrenn KD, Murphy F, Slovis CM. A toxicity study of parenteral thiamine hydrochloride. Ann Emerg Med 1989; 18: 867-70.
- Wrenn KD, Slovis CM. Is intravenous thiamine safe? Am J Emerg Med 1992; 10: 165.
- Galvin R, Brathen G, Ivashynka A, et al. EFNS guidelines for diagnosis, therapy and prevention of Wernicke encephalopathy. Eur J Neurol 2010; 17: 14-8-18.
- Giles H, Vijayan A. Fluid and Electrolyte Management. In: Green GB, Harris IS, Lin GA, Moylan KC, eds. The Washington Manual of Medical Therapeutics. 31 ed. Lippincott Williams & Wilkins, Fífadelfía, 2004: 39-71.
- Pálsson R, Guðmundsdóttir IJ, Jóhannesson AJ. Raskanir á jafnvægi vatns, elektrólýta og sýru og basa. Í: Jóhannesson AJ, Pálsson R, ritstj. Handbók í lyflæknisfræði. 3 útg. Háskólaútgáfan, Reykjavík 2006: 30-52.
- Thomson AD, Cook CC, Guerrini I, Sheedy D, Harper C, Marshall EJ. Wernicke's encephalopathy: 'Plus ca change, plus c'est la meme chose'. Alcohol Alcohol 2008; 43: 180-6.
- Ambrose ML, Bowden SC, Whelan G. Thiamin treatment and working memory function of alcohol-dependent people: preliminary findings. Alcohol Clin Exp Res 2001; 25: 112-6.