
10. tbl. 96.árg. 2010
Fræðigrein
Arfgengur skortur í ræsisameindum lektínferils komplímentvirkjunar
Inherited deficiency of the initiator molecules of the lectin-complement pathway
Ágrip
Komplímentkerfið er mikilvæg ónæmisvörn.Virkjun þess leiðir til áthúðunar og himnurofs sýkla. Þrír ferlar virkja komplímentkerfið, klassíski, styttri og lektín. Lektínferillinn er ýmist ræstur af lektínunum mannanbindilektín (MBL), fíkólín-1, fíkólín-2 eða fíkólín-3 gegnum serínpróteasa (MASP-2). Lektínin hafa svipaða byggingu og bindast sykrumynstrum á yfirborði sýkla. Erfðabreytileiki í MBL2 geninu sem veldur skorti er frekar algengur. Fjöldi rannsókna hefur sýnt að skortur er áhættuþáttur fyrir ífarandi og endurteknar sýkingar, sérstaklega þar sem aðrar ónæmisvarnir eru óþroskaðar, bældar eða gallaðar. Rannsóknir á fíkólínum eru á styttra veg komnar, en á síðasta ári var fíkólín-3-skorti lýst. Í þessu yfirliti verður fjallað um þessa ónæmisgalla sem WHO hefur nýlega skilgreint.
Inngangur
Það eru 20 ár síðan skortur á mannanbindilektíni (MBL) var fyrst tengdur við þekktan galla sermis til að miðla áthúðunarmiðluðu drápi örvera.1 Fyrstu vísbendingar um að slíkur galli væri til staðar í mannasermi komu fram árið 1968 í vísindagrein sem lýsti stúlku sem hafði þjáðst af endurteknum alvarlegum sýkingum í efri öndunarvegi fyrstu tvö ár æviskeiðs síns.2 Sjúklingurinn virtist ekki hafa neina aðra ónæmisgalla, en sermi hans var ófært um að áthúða Saccharomyces cerevisiae (bakarager). Gallinn í serminu var viðsnúanlegur þegar sermi úr öðrum einstaklingum var bætt út í. Eitthvað skorti því í sermi sjúklingsins sem aðrir virtust hafa.Tíðni áthúðunargallans meðal hvítra einstaklinga reyndist há, eða um 5-8%.3 MBL-próteinið var fyrst einangrað úr mannasermi og því lýst árið 19834 og síðar var hlutverki þess í ræsingu lektínferils komplímentkerfisins lýst.5, 6 Framfarir á sviði erfðatækninnar hafa nú leitt í ljós að MBL-skortur er arfgengur og auðvelt er að greina þá sem eru arfhreinir um genasamsætur sem leiða til skorts. Í dag hefur fjöldi rannsókna sýnt að arfgengur MBL-skortur er algengur og áhættuþáttur fyrir ýmsar sýkingar og sjálfsofnæmi. En meirihluti fólks með MBL-skort er hins vegar heilbrigður. Það er því ekki óhugsandi að mismunandi erfðabreytileikar stjórni mikilvægi og þar af leiðandi sjúkdómsmynd MBL-skorts. Á allra síðustu árum hefur verið sýnt fram á að fíkólínfjölskyldan notast við sama serínpróteasa og MBL til að ræsa komplímentkerfið. Í þessu yfirliti verður leitast við að kynna nýjustu niðurstöður í rannsóknum á þessari mikilvægu fyrstu stigs vörn mannsins gegn sýklum.
Hlutverk MBL-próteinsins
MBL þekkir og binst ákveðnum mynstrum af sykrum á yfirborði baktería, sveppa, veira og sníkjulífisfrumdýra.7 Í fyrstu var talið að hlutverk MBL væri einungis til að eyða utanaðkomandi sýklum með ræsingu komplímentkerfisins (mynd 1) en á undanförnum árum hefur komið í ljós að hlutverk MBL er mun viðameira, sérstaklega með tilliti til eyðingar á eigin frumum. MBL binst frumum í stýrðum frumudauða, frumum í vefjaskemmd (myocardial/renal/gastrointestinal ischemia reperfusion injury), mótefnasameindum, frumuleifum, frumum í æxlisvexti (ristilkrabbamein), sink-málmpróteösum, þekjufrumum í súrefnisþurrð (anoxic endothelia cells), kjarnsýrum og fósfólípíðum.8 Hlutverki MBL má því skipta í eftirfarandi tvo meginþætti: Í fyrsta lagi eru það sýklavarnir. MBL ræsir lektínferil komplímentkerfisins sem stuðlar að áthúðun sýkla sem eykur hæfni átfrumna til sýkladráps og leiðir einnig til myndunar próteinflóka (membrane-attack complex, MAC) sem rýfur bakteríuhimnur (mynd 1 og mynd 2a). Annað meginhlutverk MBL felst í stjórnun bólgusvars og viðhaldi vefja. MBL-próteinið kemur að þessum mikilvæga þætti ónæmissvars með því að stilla bólgusvörun beint (mynd 2a), stuðla að eyðingu frumna í stýrðum frumudauða (apoptosis) (mynd 2b) og stuðla að eyðingu mótefnafléttna. Því er ekki að undra að gallar í þessu mikilvæga ferli geti leitt til sjúkdóma sem einkennast af endurteknum sýkingum og/eða sjálfsofnæmistilhneigingu.
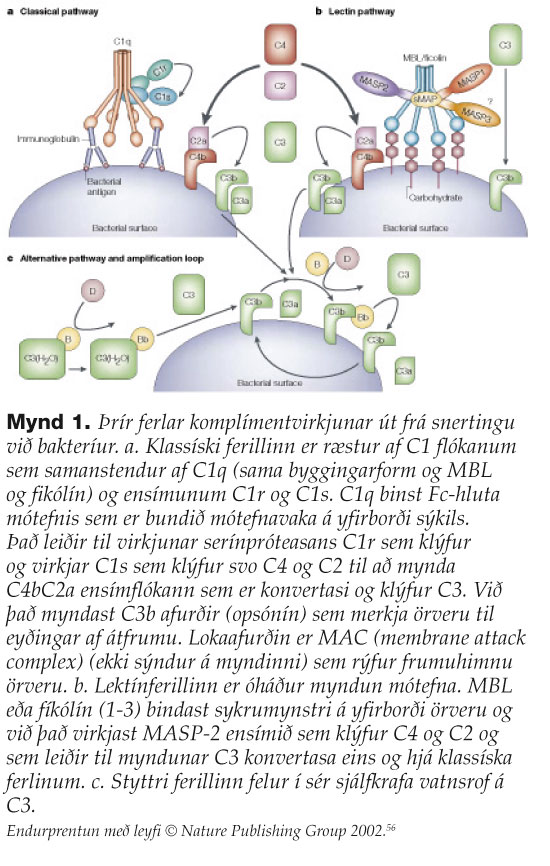{kind=link}
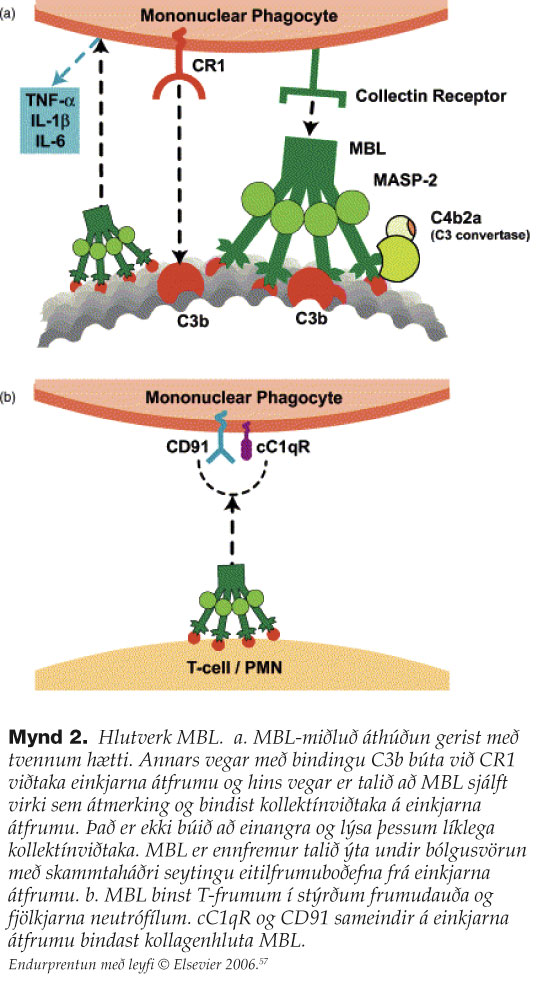{kind=link}
Sameindalíffræði MBL
MBL-próteinið finnst í sermi og er gen þess MBL2 tjáð af lifrarfrumum, en tjáning hefur einnig mælst í smágörn og eistum.9 Þrjár nákvæmlega eins fjölpeptíðkeðjur, 228 amínósýrur að lengd, eru tengdar saman og mynda undireiningu MBL-sameindarinnar7 (mynd 3). Hver keðja, sem er skráð af mismunandi útröðum MBL-2gensins, inniheldur fjögur svæði (domain): 1) 20 amínósýra cysteinríkt N-enda svæði (krosstengslasvæði) sem tekur þátt í myndun dísúlfíðtengja innan keðja og milli undireininga, 2) kollagenríkt svæði sem innheldur 18-20 endurtekningarraðir af Gly-Xaa-Yaa (tandem repeats), 3) vatnsfælið hálssvæði með αgormlaga “coiled-coil” snúningi, og 4) lektínsvæði (eða kolvetnisþekkjandi svæði). Undireiningarnar fjölliðast með dísúlfíðtengjum gegnum kollagensvæðið og mynda vöndullaga byggingu sem er líffræðilega virka formið af MBL. Algengasta form MBL í sermi eru þrí- og fjórliður. Einungis fjölliðað MBL getur ræst komplímentkerfið.
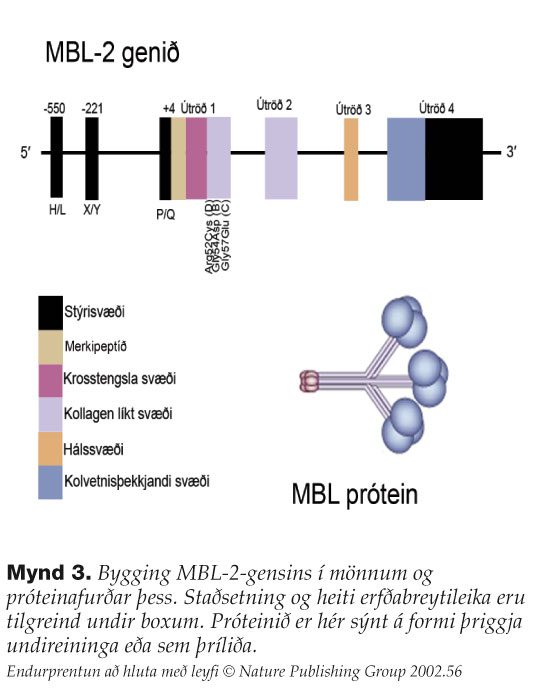{kind=link}
MBL2 genið er staðsett á litningi 10 (q11.2-q21) og samanstendur af fjórum útröðum (mynd 3). Erfðabreytileika eða SNP (single nucleotide polymorphism) hefur verið lýst í geninu. Í útröð eitt finnast þrjár algengar punktstökkbreytingar staðsettar í tákna 54, 57 og 52, einnig þekktar sem B, C og Dstökkbrigði (variant alleles) (mynd 3)10. Stökkbreyting B er G®A breyting sem veldur Gly ®Asp breytingu í fimmtu endurtekningarröðinni Gly-Xaa-Yaa.11 Stökkbreyting C er G®A breyting sem veldur Gly®Asp breytingu í sjöttu endurtekningarröðinni Gly-Xaa-Yaa.12 Stökkbreyting D er C®T breyting sem veldur Cys®Arg breytingu.13 B, C og Dstökkbrigðin sýna álíka svipgerð og því eru þau ekki aðgreind og kölluð O. Arfhreinir eða arfblendnir einstaklingar um útraðastökkbrigðin eru því með arfgerð O/O (B/B, B/D, D/D og svo framvegis). Villigerðarsamsæta í útröð kallast A til aðgreiningar frá útraðastökkbrigðunum. Að auki er til staðar erfðabreytileiki í stýrisvæði MBL2 í stöðu -550 (H/L), -221 (Y/X) og +4 (P/Q) (mynd 3).14
Áhrif erfðabreytileika í MBL-2 geninu á styrk MBL í sermi
Einstaklingar með arfhreina villigerð eða A/A eru almennt með MBL-styrk hærri en 1000 ng/ml (mynd 4).15 Útraðastökkbrigðin (O) valda röskun í fjölliðun MBL sem leiðir af sér óstöðugt og óvirkt prótein.16-18 Stökkbreytt MBL hefur skertan eiginleika til að bindast bindlum sínum og þar af leiðandi er lektínferilmiðluð ræsing komplímentkerfisins óskilvirk eða löskuð. Einstaklingar sem eru arfblendnir (A/O) hafa marktækt lægri MBL-styrk, sé miðað við meðalstyrk arfhreinnar villigerðar, og eru yfirleitt á bilinu 500-1000 ng/ml (mynd 4).19 O/Oeinstaklingar eru með MBL-styrk í sermi minni en 50 ng/ml sem eru neðstu mælingarmörk (detection limit) flestra styrksprófa (mynd 4).
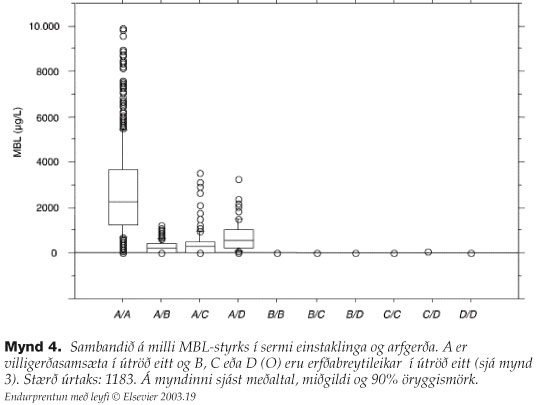{kind=link}
Erfðabreytileiki í stýrisvæði hefur áhrif á tjáningu MBL2 gensinsog ákvarðar styrk MBL í sermi einstaklings.20 Einstaklingar með HY, LY eða LX erfðabreytileika eru með háan, meðal eða lágan MBL-styrk.14 Erfðabreytileikarnir í útröð og stýrisvæði eru í tengslaójafnvægi og hefur aðeins sjö algengum haplótýpum verið lýst, HYPA, LYPA, LYQA, LXPA, LYQC, LYPB og HYPD.21
Skilgreining á MBL-skorti
Styrkur MBL í sermi er mjög misjafn milli einstaklinga en hins vegar stöðugur hjá hverjum og einum ævilangt. Styrksbilið nær frá 5 ng til meira en 10 µg á millilítra.15 Það verður þreföld aukning í MBL-styrk í bráðafasa bólguviðbrögðum.22 Mismunandi MBL-styrkur er þó fyrst og fremst tilkominn vegna erfðabreytileika og í því tilfelli getur munurinn verið allt að þúsundfaldur.
Skilgreiningin á MBL-skorti og viðmiðunarmörk hafa verið misjafnlega útfærð í rannsóknum og því hefur reynst erfitt að bera niðurstöður milli rannsókna saman. Í nýlegri „meta-analysu“ grein eru viðmiðunarmörkin á kerfisbundinn hátt skilgreind sem 500 ng/ml út frá 1642 heilbrigðum einstaklingum úr fjórum mismunandi þýðum, þar með talið íslensku þýði.23 Einnig var sýnt fram á að einstaklingar með XA/O og O/O arfgerðir hefðu lægsta MBL-styrk í sermi, eða minna en 50 ng/ml. X er erfðabreytileiki í stýrisvæði MBL2 gensins sem veldur lágri tjáningu á MBL2 geninu (sjá kaflann hér á undan).
Algengi MBL-skorts
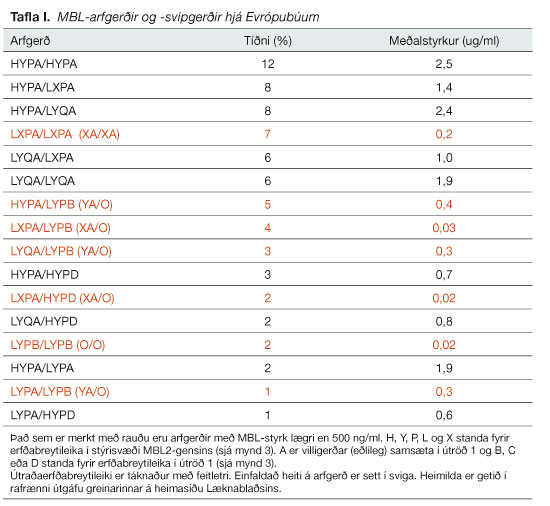
Í töflu I má sjá algengi mismunandi arfgerða hjá Evrópubúum sem hafa MBL-styrk lægri en 500 ng/ml (litað með rauðu). Um 24% Evrópubúa eru með MBL-styrk neðan við 500 ng/ml. Þar af eru 8% einstaklinga með MBL-styrk lægri en 50 ng/ml. Innan þessa hóps eru einungis arfgerðirnar XA/O og O/O sem í dag eru nefndar lágstyrks- eða skortsarfgerðir MBL (sjá líka kafla hér á undan).
{kind=link}
Erfðabreytileiki í MASP-2 geninu og MASP2-skortur
MBL myndar flóka við þrjá mismunandi serínpróteasa, (MASP: MBL-associated serine proteases) MASP-1, MASP-2 og MASP-3 (mynd 1).5, 24, 25 Að auki inniheldur virkur MBL-MASP flókinn lítið MASP-prótein (sMAP eða MAp19) sem hefur enga serínpróteasa virkni.26, 27 MASP-2 er samsvarandi við C1s ensím klassíska ferilsins vegna þess að það klýfur C4 og C2 (mynd 1).5 Þegar MBL binst yfirborðssykrum virkjast MASP-2 sem klýfur C4 og C2 til myndunar á C3 konvertasa (C4b2a) (mynd 1). MASP-2 binst einnig sameindunum fíkólín-1, fíkólín-2 og fíkólín-3, sem líkt og MBL þekkja ýmis sykurmynstur á örverum og ræsa lektínferilinn (sjá síðar).
Nýlega var þremur erfðabreytileikum (R99Q, D120G og V377A) lýst í MASP-2 geninu sem er staðsett á litningi 1p36.2-3.28-31 Tíðni stökkbreyt-inganna í úrtaki hvítra Dana er 0,14% fyrir R99Q, 3,9 % fyrir D120G og 1% fyrir V377A.31 Fleiri erfðabreytileikum hefur verið lýst, sem finnast ekki hjá hvítum kynþætti. Sýnt hefur verið fram á in vitro að raðbrigði (recombinant) af D120G MASP-2 getur ekki bundist MBL og ekki klofið C4 og þar af leiðandi ekki virkjað komplímentkerfið.28, 32 Raðbrigði af R99Q og V377A MASP-2 voru hins vegar ekki frábrugðin raðbrigðavilligerð með tilliti til virkjunar komplímentkerfisins. Athyglisvert er að árið 2003 var 36 ára gömlum einstaklingi lýst, sem var með óútskýrðar endurteknar sýkingar og krónískar bólgur og kom í ljós að hann var arfhreinn fyrir D120G stökkbrigðið og ekkert MASP-2 prótein mældist í sermi hans.28 Þessi einstaklingur var hins vegar með háan MBL-styrk í blóði en samt gat sermi hans ekki virkjað lektínferilinn, sem líklega má rekja til MASP-2-skortsins. Það mætti því ef til vill halda því fram að MASP-2 skortur geti haft víðtækari áhrif en MBL-skortur einn og sér, því að MASP-2 er ekki bara ábyrgt fyrir líffræðilegri virkni MBL heldur einnig virkni fíkólína (mynd 1).
Hér var því talið líklegt að um áður óþekktan ónæmisgalla væri að ræða. Síðan árið 2003 hefur verið skimað fyrir þessum erfðabreytileika í mismunandi sjúklingahóprannsóknum. Sex arfhreinir (D120G/D120G) einstaklingar hafa verið greindir. Fjórir af þeim voru börn sem komu úr mismunandi sjúklingahópum. Eitt barn kom úr barnahópi sem var með endurteknar öndunarfærasýkingar,33 eitt barn úr psoriasishópi,34 eitt úr hópi með slímseigjusjúkdóm (cystic fibrosis) (barn með mjög alvarlegan lungnasjúkdóm)35 og eitt barn úr barnahópi með endurteknar sýkingar.36 Tveir af þessum sex einstaklingum sem hafa verið greindir arfhreinir, voru fullorðnir og komu úr hópi heilbrigðs viðmiðunarþýðis (tvær heilbrigðar konur um fertugt).37 Sermi arfhreinna einstaklinga getur ekki virkjað komplímentkerfið gegnum lektínferilinn (mælt með virkniprófi, sjá hér að neðan) og samkvæmt niðurstöðum þessara rannsókna geta fullorðnir verið fullkomlega heilbrigðir án starfhæfs lektínferils. Áframhaldandi skimanir á mismunandi sjúklingahópum mun líklega gefa meiri upplýsingar og skilning á mikil
vægi lektínferilsins. Einnig er mögulegt að eitthvað annað þekkt eða óþekkt ónæmiskerfi komi í stað lektínferilsins hjá þeim sem eru með á skortMBL eða MASP2. Tíðni arfhreinna D120G einstaklinga hefur verið áætluð sem 1/1000.38
Erfðabreytileiki í genum fíkólína
Bygging fíkólína er mjög lík byggingu MBL. Í stað kolvetnisþekkjandi svæða hafa þau fibrínógen-lík svæði.39 Þau starfa líkt og MBL, það er þau ræsa lektínferilinn gegnum MASP-2 ensímið (mynd 1). Af hinum fjórum ræsisameindum er fíkólín-3-styrkur í sermi langhæstur, eða 25 µg/ml, síðan kemur fíkólín-2 styrkur (5 µg/ml), þá MBL (1 µg/ml) og síðast fíkólín-1 (<0,1µg/ml).40 Í mönnum er fíkólín-3 mRNA aðallega tjáð í lungum og talsvert í lifur.40 Fíkólín-2 mRNA er tjáð aðallega í lifur en lítillega í beinmerg, hálskirtlum og görn.40 Á hinn bóginn finnst fíkólín-1 mRNA ekki í lifur heldur aðallega í hvítum blóðfrumum (leukocytes) og beinmerg en lítillega í milta og lungum.40
Á síðasta ári birtist grein sem lýsir í fyrsta sinn einstaklingi með fíkólín-3-skort.41 Hann hafði verið með endurteknar sýkingar frá unga aldri, þar á meðal alvarlegar sýkingar í neðri öndunarvegi, sýkingu í heila (bilateral frontal cereberal abscesses) af völdum óhemólýtískra streptókokka og vörtur á fingrum. Aðrir ferlar ónæmiskerfis störfuðu innan eðlilegra marka samkvæmt mælingum, en hins vegar var fíkólín-3 í blóði ekki mælanlegt. Fíkólín-3 er tjáð af FCN3 geninu og er tjáð í lungum og lifur. Þegar viðkomandi var arfgerðargreindur kom fram hugsanleg skýring á sjúkleika hans. Hann var arfhreinn um stökkbreytingu (1637C) sem veldur breytingu í lesramma FCN3 gensins og gefur af sér óstarfhæft og gallað fíkólín-3. Ekki var lýst sjúkrasögu arfblendinna einstaklinga en styrkur fíkólín-3 í blóði þeirra var um 50% af villigerð. Tíðni arfblendinna meðal hvítra einstaklinga er 1,8%.41, 42
Það er mjög sennilegt að vitneskja okkar um hlutverk og mikilvægi fíkólína í ræsingu lektínferilsins fari stigvaxandi á næstu árum. Því telja greinarhöfundar mikilvægt að læknar geri sér grein fyrir tilurð þessara nýskilgreindu ónæmisgalla meðal einstaklinga með endurteknar sýkingar eins og að ofan greinir.
MBL-skortur metinn út frá virkni lektínferilsins
Á ónæmisfræðideild Landspítala er í dag boðið upp á að mæla MBL-styrk í sermi einstaklinga. Prófið var þróað af deildinni út frá aðferð Claus Kock við Statens Serum Institut í Danmörku og byggist á samloku-ELISA-aðferð (Ensyme-Linked Immunosorbent Assay) þar sem húðað er með mótefni gegn MBL.43 Neðstu greiningarmörk mælingarinnar eru 20 ng/ml.
Eftir því sem meira er vitað um MBL og samverkandi sameindir þess virðist MBL-styrksmæling ekki gefa fullnægjandi svör um starfsemi lektínferilsins hjá sjúklingum. MBL-styrksmæling mælir ekki MASP-2-skort (sjá kafla um MASP-2-skort hér að ofan). Áætlað er að ónæmisfræðideildin bjóði framvegis upp á próf sem mælir skilvirkni lektínferilsins. Prófið er vottuð ELISA-aðferð þar sem plötur eru húðaðar með sykrunni mannan og því er MBL-miðluð ræsing sérhæft mæld.44 Þar sem fíkólín bindast ekki mannan er ekki verið að mæla ræsingu af þeirra völdum. Í mælingunni er komið í veg fyrir ræsingu klassíska ferilsins með því að bæta út í C1q-hindra. Prófið mælir lokaafurð komplímentræsingar eða MAC (membrane attack complex) (C5b-9). Niðurstöður eru gefnar upp sem prósentur af viðmiði. Viðmiðunargildi fyrir virkniskort eru <10% virkni samkvæmt mælingunni. Áætlað er að nota virkniprófið sem skimpróf og ef einstaklingur mælist undir viðmiðunarmörkum er hægt að staðfesta með styrk og/eða arfgerð. Stöðluð virknipróf sem mæla fíkólínmiðlaða ræsingu eru enn ekki fáanleg en væntanlega munu þau koma fljótlega á markað og líklegt er að ónæmisfræðideild taki þau í notkun fyrir rútínumælingar og vísindarannsóknir.
Við gæðaprófun á ofangreindu virkniprófi hjá ónæmisfræðideild voru 130 manns með þekktan MBL-styrk mældir. Það var línulegt samband milli styrks og virkni en þó voru undantekningar. Sex manns sem áður höfðu mælst með MBL-styrk <500 ng/ml, mældust með yfir 50% virkni, þar af einn með 100% virkni. Það er mjög sennilegt að þessir einstaklingar hafi XA/XA arfgerð (lágur styrkur en virkt prótein). Ætlunin er að nota virkniprófið til að bera virkni lektínferilsins hjá XA/XAeinstaklingum saman við virknina hjá O/O einstaklingum (lágur styrkur og gallað prótein).
Tengsl MBL-skorts við sjúkdóma
Fjöldi rannsókna hefur verið gerður til að finna möguleg tengsl MBL-skorts við sjúkdóma. Helstu rannsóknir þar sem tengsl hafa fundist eru teknar saman í töflu II. Til að gæta samræmis eru hér aðeins teknar saman rannsóknir þar sem miðað er við tengsl sjúkdóma við skortsarfgerðirnar XA/O og O/O. Einstaklingar með þessar arfgerðir geta verið heilbrigðir og almennt er talið að aðrir meðfæddir ónæmisgallar eða ónæmisbæling þurfi að vera til staðar til að MBL-skortur hafi einhverja klíníska þýðingu.45 Þó hafa nýlegar rannsóknir sýnt fram á hærri tíðni MBL-skortsarfgerða hjá fullorðnum með alvarlegar, sjaldgæfar og endurteknar sýkingar, óháð því hvort þeir eru með meðfædda ónæmisgalla.46 Sjá nánar í töflu II. Á undanförnum árum hafa tengsl MASP2-skorts við sjúkdóma/sjúklingahópa einnig verið rannsökuð en ekki verður fjallað um þær niðurstöður í þessu yfirliti. Þess ber að geta að MBL-skortur og MASP-2-skortur hafa nýlega verið flokkaðir sem ónæmisgallar (primary immunodeficiencies (PID)).47
{kind=link}
Dýralíkön af MBL-skorti
Mýs eru með tvö gen (MBL-A og –C) sem skrá fyrir öðruvísi MBL-sameindum en manna. Í „sepsis“ músarlíkani, þar sem Staphylococcus aureus var sprautað í mýs með úrfelld MBL- gen, var sýnt fram á mikilvægi MBL gegn bakteríunni, því að allar erfðabreyttu mýsnar dóu samanborið við 55% lifun villigerðarmúsa.48 Hægt var að bjarga stökkbreyttu músunum með því gefa þeim raðbrigða-MBL-prótein í æð. Í sama líkani hefur verið sýnt fram á marktækt minna viðnám gegn herpes simplex veiru 2.49 Engin einkenni sjálfsofnæmissjúkdóma fundust í músunum en hins vegar var skertur eiginleiki til að hreinsa upp frumur í stýrðum frumudauða.50 In vivo genalækning með MBLA-kjarnsýrum verndaði ónæmisbældar mýs sem í var grætt ristilkrabbameinsfrumulína sem sýnir að músa-MBL getur hamlað æxlisvexti.51 MBL-skortur er hins vegar verndandi í músum með blóðþurrð (ischaemia/reperfusion injury) í hjarta, lifur og meltingarvegi.52 Þessar niðurstöður eru í samræmi við nýlega klíníska rannsókn í mönnum þar sem haplótýpan LYQA (hár MBL-styrkur í blóði) var metin sem áhættuþáttur fyrir drep í hjartavöðva hjá sjúklingum sem hafa gengist undir kransæðaaðgerð.53
Lokaorð
Nú eru liðin 20 ár síðan því var fyrst lýst að skert geta sermis til áthúðunar væri tengd sameindinni MBL eða réttara sagt skorti á henni.1 MBL er þróunarfræðilega varðveitt sameind og hlýtur því að gegna mikilvægu hlutverki. Ofangreindar rannsóknaniðurstöður sýna ótvírætt fram á að ræsing lektínferilsins gegnir lykilhlutverki í ósértæku ónæmissvari og viðheldur heilbrigði. Margt bendir eindregið til þess að meðfæddir gallar í ferlinu séu algengari og klínískt mikilvægi þeirra meira en talið hefur verið. Lítið hefur verið rannsakað hvernig og hvort MBL-skortur sé bættur upp, til dæmis af fíkólínum, en greinarhöfundar hyggjast rannsaka það nánar.
Mikil þróunarvinna hefur verið unnin á undanförnum árum á notkun MBL-próteinsins hjá einstaklingum með skort og var ein slík rannsókn framkvæmd á ónæmisfræðideild Landspítala.54, 55 Þó enn sé langt í land með að skilgreina meðferðarleiðir er þó hugsanlegt að bráðum muni ákveðnum áhættuhópum standa slík meðferð til boða.
Heimildir
- Super M, Thiel S, Lu J, et al. Association of low levels of mannan-binding protein with a common defect of opsonisation. Lancet 1989; 2: 1236-9.
- Miller ME. A familial, plasma-associated defect of phagocytosis. Lancet 1968 (ii): 60-3.
- Soothill JF, Harvey BA. Defective opsonization. A common immunity deficiency. Arch Dis Child 1976; 51: 91-9.
- Kawasaki N, Kawasaki T, Yamashina I. Isolation and characterization of a mannan-binding protein from human serum. J Biochem 1983; 94: 937-47.
- Thiel S, Vorup-Jensen T, Stover CM, et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature 1997; 386: 506-10.
- Ikeda K, Sannoh T, Kawasaki N, et al. Serum lectin with known structure activates complement through the classical pathway. J Biol Chem 1987; 262: 7451-4.
- Dommett RM, Klein N, Turner MW. Mannose-binding lectin in innate immunity: past, present and future. Tissue Antigens 2006; 68: 193-209.
- Takahashi K, Ip WE, Michelow IC, et al. The mannose-binding lectin: a prototypic pattern recognition molecule. Curr Opin Immunol 2006; 18: 16-23.
- Seyfarth J, Garred P, Madsen HO. Extra-hepatic transcription of the human mannose-binding lectin gene (mbl2) and the MBL-associated serine protease 1-3 genes. Mol Immunol 2006; 43: 962-71.
- Sumiya M, Super M, Tabona P, et al. Molecular basis of opsonic defect in immunodeficient children. Lancet 1991; 337: 1569-70.
- Heise CT, Nicholls JR, Leamy CE, et al. Impaired secretion of rat mannose-binding protein resulting from mutations in the collagen-like domain. J Immunol 2000; 165: 1403-9.
- Lipscombe RJ, Sumiya M, Hill AV, et al. High frequencies in African and non-African populations of independent mutations in the mannose binding protein gene. Hum Mol Genet 1992; 1: 709-15.
- Madsen HO, Garred P, Kurtzhals JA, et al. A new frequent allele is the missing link in the structural polymorphism of the human mannan-binding protein. Immunogenetics 1994; 40: 37-44.
- Madsen HO, Garred P, Thiel S, et al. Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J Immunol 1995; 155: 3013-20.
- Thiel S, Frederiksen PD, Jensenius JC. Clinical manifestations of mannan-binding lectin deficiency. Mol Immunol 2006; 43: 86-96.
- Larsen F, Madsen HO, Sim RB, et al. Disease-associated mutations in human mannose-binding lectin compromise oligomerization and activity of the final protein. J Biol Chem 2004; 279: 21302-11.
- Wallis R, Cheng JY. Molecular defects in variant forms of mannose-binding protein associated with immunodeficiency. J Immunol 1999; 163: 4953-9.
- Wallis R. Dominant effects of mutations in the collagenous domain of mannose-binding protein. J Immunol 2002; 168: 4553-8.
- Garred P, Larsen F, Madsen HO, et al. Mannose-binding lectin deficiency--revisited. Mol Immunol 2003; 40: 73-84.
- Naito H, Ikeda A, Hasegawa K, et al. Characterization of human serum mannan-binding protein promoter. J Biochem 1999; 126: 1004-12.
- Garred P, Larsen F, Seyfarth J, et al. Mannose-binding lectin and its genetic variants. Genes Immun 2006; 7: 85-94.
- Thiel S, Holmskov U, Hviid L, et al. The concentration of the C-type lectin, mannan-binding protein, in human plasma increases during an acute phase response. Clin Exp Immunol 1992; 90: 31-5.
- Eisen DP, Dean MM, Boermeester MA, et al. Low serum mannose-binding lectin level increases the risk of death due to pneumococcal infection. Clin Infect Dis 2008; 47: 510-6.
- Dahl MR, Thiel S, Matsushita M, et al. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity 2001; 15: 127-35.
- Sato T, Endo Y, Matsushita M, et al. Molecular characterization of a novel serine protease involved in activation of the complement system by mannose-binding protein. Int Immunol 1994; 6: 665-9.
- Stover CM, Thiel S, Thelen M, et al. Two constituents of the initiation complex of the mannan-binding lectin activation pathway of complement are encoded by a single structural gene. J Immunol 1999; 162: 3481-90.
- Takahashi M, Endo Y, Fujita T, et al. A truncated form of mannose-binding lectin-associated serine protease (MASP)-2 expressed by alternative polyadenylation is a component of the lectin complement pathway. Int Immunol 1999; 11: 859-63.
- Stengaard-Pedersen K, Thiel S, Gadjeva M, et al. Inherited deficiency of mannan-binding lectin-associated serine protease 2. N Engl J Med 2003; 349: 554-60.
- Lozano F, Suarez B, Munoz A, et al. Novel MASP2 variants detected among North African and Sub-Saharan individuals. Tissue Antigens 2005; 66: 131-5.
- Stover C, Endo Y, Takahashi M, et al. The human gene for mannan-binding lectin-associated serine protease-2 (MASP-2), the effector component of the lectin route of complement activation, is part of a tightly linked gene cluster on chromosome 1p36.2-3. Genes Immun 2001; 2: 119-27.
- Thiel S, Steffensen R, Christensen IJ, et al. Deficiency of mannan-binding lectin associated serine protease-2 due to missense polymorphisms. Genes Immun 2007; 8: 154-63.
- Thiel S, Kolev M, Degn S, et al. Polymorphisms in mannan-binding lectin (MBL)-associated serine protease 2 affect stability, binding to MBL, and enzymatic activity. J Immunol 2009; 182: 2939-47.
- Cedzynski M, Szemraj J, Swierzko AS, et al. Mannan-binding lectin insufficiency in children with recurrent infections of the respiratory system. Clin Exp Immunol 2004; 136: 304-11.
- Stover C, Barrett S, Lynch NJ, et al. Functional MASP2 single nucleotide polymorphism plays no role in psoriasis. Br J Dermatol 2005; 152: 1313-5.
- Olesen HV, Jensenius JC, Steffensen R, et al. The mannan-binding lectin pathway and lung disease in cystic fibrosis--disfunction of mannan-binding lectin-associated serine protease 2 (MASP-2) may be a major modifier. Clin Immunol 2006; 121: 324-31.
- Cedzynski M, Atkinson AP, St Swierzko A, et al. L-ficolin (ficolin-2) insufficiency is associated with combined allergic and infectious respiratory disease in children. Mol Immunol 2009; 47: 415-9.
- Garcia-Laorden MI, Sole-Violan J, Rodriguez de Castro F, et al. Mannose-binding lectin and mannose-binding lectin-associated serine protease 2 in susceptibility, severity, and outcome of pneumonia in adults. J Allergy Clin Immunol 2008; 122: 368-74, 374 e1-2.
- Sorensen R, Thiel S, Jensenius JC. Mannan-binding-lectin-associated serine proteases, characteristics and disease associations. Springer Semin Immunopathol 2005; 27: 299-319.
- Endo Y, Matsushita M, Fujita T. Role of ficolin in innate immunity and its molecular basis. Immunobiology 2007; 212: 371-9.
- Garred P, Honore C, Ma YJ, et al. MBL2, FCN1, FCN2 and FCN3-The genes behind the initiation of the lectin pathway of complement. Mol Immunol 2009; 46: 2737-44.
- Munthe-Fog L, Hummelshoj T, Honore C, et al. Immunodeficiency associated with FCN3 mutation and ficolin-3 deficiency. N Engl J Med 2009; 360: 2637-44.
- Munthe-Fog L, Hummelshoj T, Ma YJ, et al. Characterization of a polymorphism in the coding sequence of FCN3 resulting in a Ficolin-3 (Hakata antigen) deficiency state. Mol Immunol 2008; 45: 2660-6.
- Saevarsdottir S, Vikingsdottir T, Vikingsson A, et al. Low mannose binding lectin predicts poor prognosis in patients with early rheumatoid arthritis. A prospective study. J Rheumatol 2001; 28: 728-34.
- Seelen MA, Roos A, Wieslander J, et al. Functional analysis of the classical, alternative, and MBL pathways of the complement system: standardization and validation of a simple ELISA. J Immunol Methods 2005; 296: 187-98.
- Aittoniemi J, Baer M, Soppi E, et al. Mannan binding lectin deficiency and concomitant immunodefects. Arch Dis Child 1998; 78: 245-8.
- Hoeflich C, Unterwalder N, Schuett S, et al. Clinical manifestation of mannose-binding lectin deficiency in adults independent of concomitant immunodeficiency. Hum Immunol 2009; 70: 809-12.
- Notarangelo L, Casanova JL, Conley ME, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee Meeting in Budapest, 2005. J Allergy Clin Immunol 2006; 117: 883-96.
- Shi L, Takahashi K, Dundee J, et al. Mannose-binding lectin-deficient mice are susceptible to infection with Staphylococcus aureus. J Exp Med 2004; 199: 1379-90.
- Gadjeva M, Paludan SR, Thiel S, et al. Mannan-binding lectin modulates the response to HSV-2 infection. Clin Exp Immunol 2004; 138: 304-11.
- Stuart LM, Takahashi K, Shi L, et al. Mannose-binding lectin-deficient mice display defective apoptotic cell clearance but no autoimmune phenotype. J Immunol 2005; 174: 3220-6.
- Ma Y, Uemura K, Oka S, et al. Antitumor activity of mannan-binding protein in vivo as revealed by a virus expression system: mannan-binding proteindependent cell-mediated cytotoxicity. Proc Natl Acad Sci U S A 1999; 96: 371-5.
- Takahashi K. Lessons learned from murine models of mannose-binding lectin deficiency. Biochem Soc Trans 2008; 36: 1487-90.
- Collard CD, Shernan SK, Fox AA, et al. The MBL2 'LYQA secretor' haplotype is an independent predictor of postoperative myocardial infarction in whites undergoing coronary artery bypass graft surgery. Circulation 2007; 116(11 Suppl): I106-12.
- Valdimarsson H, Vikingsdottir T, Bang P, et al. Human plasma-derived mannose-binding lectin: a phase I safety and pharmacokinetic study. Scand J Immunol 2004; 59: 97-102.
- Valdimarsson H. Infusion of plasma-derived mannan-binding lectin (MBL) into MBL-deficient humans. Biochem Soc Trans 2003; 31: 768-9.
- Fujita T. Evolution of the lectin-complement pathway and its role in innate immunity. Nat Rev Immunol 2002; 2: 346-53.
- Turner MW. The role of mannose-binding lectin in health and disease. Mol Immunol 2003; 40: 423-9.