
05. tbl. 96.árg. 2010
Fræðigrein
Sjúkratilfelli - lífshættulegar truflanir á blóðsöltum hjá átta vikna dreng
A case report – Severe electrolyte disturbances in an eight week old boy
Ágrip
Natríumskortur er algengasta salttruflun sem kemur fyrir hjá börnum og geta einkenni verið mjög mismunandi, allt frá einkennaleysi til alvarlegra einkenna frá miðtaugakerfi. Algengasta orsök natríumskorts er tap á vökva og natríum um meltingarveg en sé tapið um nýru getur orsökin meðal annars verið skortur á saltsteranum aldósteróni sem hefur megin hlutverk í stýringu saltjafnvægis líkamans. Þegar virkni aldósteróns vantar verður minni seyting á kalíum og vetnisjónum í nýrnapíplum auk þess sem endurupptaka á natríum verður minni og natríumskortur hlýst af. Kalíumofgnótt, sem getur valdið lífshættulegum hjartsláttartruflunum, er einnig einkennandi fyrir lága virkni aldósteróns. Algengasta orsök fyrir minnkaðri myndun aldósteróns er skortur á ensíminu 21-hydroxylasa (21-OH) sem veldur sjúkdómnum congenital adrenal hyperplasia (CAH) en svipuð einkenni geta komið fram í sjaldgæfum sjúkdómi, pseudohýpóaldósterónismus (PHA), þar sem truflun er í virkni aldósteróns. Hér er fjallað um dreng með lífshættulegar brenglanir á blóðsöltum vegna PHA sem orsakaðist af þvagfærasýkingu og bakflæði.
Sjúkrasaga
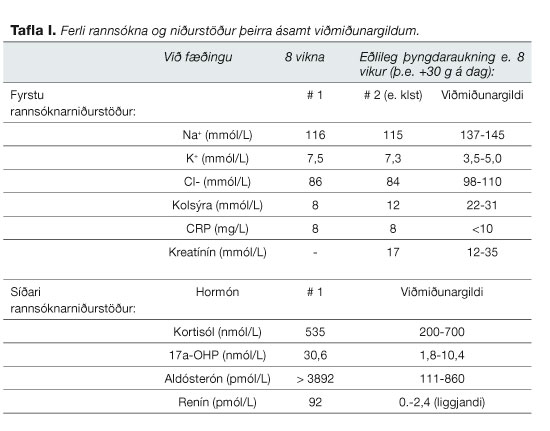
Vegna gruns um hjartagalla var átta vikna dreng vísað á Barnaspítala Hringsins. Hann hafði þyngst illa og hafði nýlega byrjað að kasta upp. Skoðun var ómarkverð fyrir utan að hann var dálítið gugginn og með bláma á útlimum, hiti 37,1ºC. Meðganga og fæðing hafði verið eðlileg en hann hafði einungis þyngst um 600 grömm frá fæðingu. Bráð hjartaómun og röntgenmynd af brjóstholi leiddu ekkert óeðlilegt í ljós. Fjölskyldusaga leiddi ekkert markvert í ljós. Blóðprufur sýndu fram á verulegan natríumskort, kalíumofgnótt og lága kolsýru (sjá töflu I). Blóðstatus og CRP voru eðlileg en hvít blóðkorn sáust í þvagprufu. Einnig voru teknar blóðprufur til hormónamælinga.
Vegna natríumskorts og kalíumofgnóttar vaknaði grunur um congenital adrenal hýperplasiu (CAH). Sem fyrsta meðferð var gefið hratt innrennsli af saltvatni (20 mL/kg á tveimur klst), sýklalyf í æð (Zinacef®) og hleðsluskammtur af sykur- og saltsterum. Á meðan drengurinn var í meðferð og eftirliti á bráðamóttöku fékk hann skyndilegt slappleikakast og hjartarafrit sýndi slegla-
hraðtakt sem hægt var að stöðva með líkamlegri örvun. Drengurinn var lagður inn á gjörgæslu til eftirlits og síðar á barnadeild til frekari rannsókna.
Nokkrum dögum eftir innlögn bárust niðurstöður hormónamælinga og var þá ljóst að vinnugreiningin, salttapandi CAH, gat ekki staðist. Kortisól mældist eðlilegt og 17a-hýdroxýprógesterón (17-OHP) mældist einungis vægt hækkað (tafla I). Aldósterón reyndist verulega hækkað sem og renín (tafla I). Þessar mæl-ingar útilokuðu ennfremur skort á aldósterón synþetasa. Í kjölfarið var allri steragjöf hætt.
{kind=link}
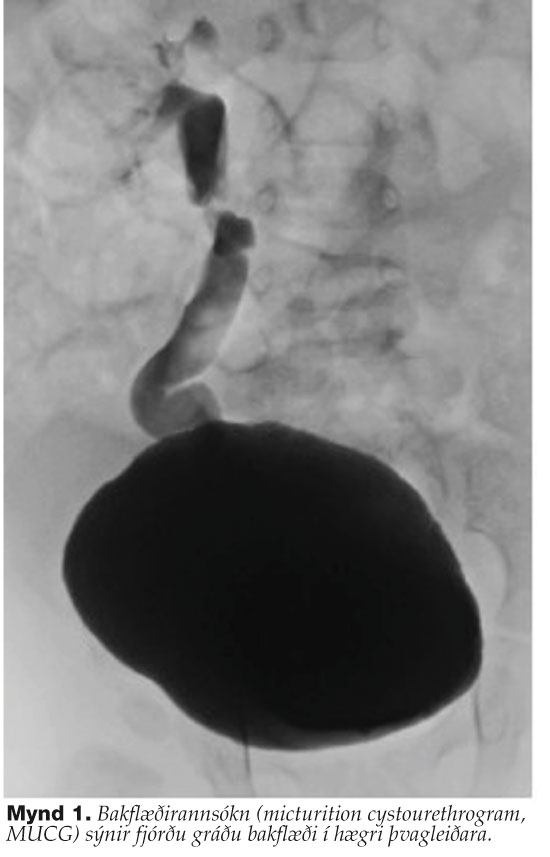
Á fimmta degi innlagnar var þvagfærasýking staðfest með jákvæðri ræktun E. coli með góðu næmi fyrir sýklalyfjum. Bakflæðirannsókn (micturition cystourethrogram, MUCG) sýndi fjórðu gráðu bakflæði bæði passíft og aktíft hægra megin (mynd 1). Technetium-99m merkt dímerkaptósúksíník sýra (Technetium-99m-labeled dimercaptosuccinic acid, DSMA) ísótópaskann af nýrum sýndi mjög ósamhverfa upphleðslu í nýrum, 79% í því vinstra en 21% í því hægra auk misdreifðrar upphleðslu. Við speglun á þvagblöðru sást að þvagleiðaraop voru eðlilega staðsett en bæði stór og víð, það hægra áberandi meira. Ný klínísk
greining var því pseudóhypóaldósterónismus (PHA), hugsanlega orsakaður af þvagfærasýkingu. Einkenni og brenglanir á blóðsöltum gengu að fullu til baka. Ákveðið var að fylgjast náið með drengnum áfram og hafa hann á fyrirbyggjandi sýklalyfjum.
{kind=link}
Rúmu ári síðar dafnaði drengurinn vel og leið vel. Blóðsölt mældust eðlileg og virtist aldósterón hafa náð jafnvægi en síðasta mæling var innan eðlilegra marka. Áður hafði aldósterón mælst ítrekað verulega hækkað eða á bilinu 1500-2000 pmól/L. Ekki hefur fundist stökkbreyting í geni sem kóðar fyrir saltsteraviðtakann (e. mineralocorticoid receptor, MR). Dagleg sýklalyfjagjöf (Primazol (8 + 40 mg/ml) 2ml á dag) hefur komið í veg fyrir sýkingar.
Umræður
Hér er sagt frá dreng með lífshættulegar brenglanir á blóðsöltum orsakaðar af þvagfærasýkingu og bakflæði. Þetta sjúkdómstilfelli er mikilvæg áminning um að alvarleg þvagfærasýking hjá ungum börnum getur verið einkennalítil. Að auki sýnir dæmið gildi þess að athuga blóðsölt hjá börnum með óljós einkenni.
Natríumþéttni í blóði er stýrt í nýrum með mismunandi frásogi á natríum og vatni í þvagi. Natríumskortur er algengasta saltröskunin og finnst hjá allt að 3% barna sem lögð eru inn á spítala.1 Einkenni natríumskorts eru mjög einstaklingsbundin og fara bæði eftir því hversu lágt natríumgildið er og hversu hröð lækkunin er en í flestum tilfellum er talið að einkenni komi fram við 125 mmól/L. Einkenni geta verið frá miðtaugakerfi, vöðvum og hjarta- og æðakerfi. Lækkun á natríum í blóði leiðir til aukins osmósustiguls yfir blóðheilaþröskuld og heilabjúgur getur myndast. Einkenni eins og höfuðverkur, ógleði, uppköst, pirringur eða flog koma til vegna aukins innankúpuþrýstings. Hlutfall heilastærðar og kúpurýmis er stærra hjá börnum miðað við fullorðna. Heili barns nær fullorðinsstærð við sex ára aldur en fullorðinsstærð kúpu næst ekki fyrr en við 16 ára aldur. Af þessum sökum eru börn í meiri hættu á að þróa með sér einkenni eða heilamein í tengslum við natríumskort.2,3
Lækkuð natríumþéttni (Na+ <130 mmól/L í sermi) getur orsakast á þrjá mismunandi vegu: 1) ofmagn vökva, 2) of mikill útskilnaður natríum, annaðhvort um nýru eða annars staðar frá og 3) ónóg inntaka á natríum. Algengasta orsök natríumskorts hjá börnum er vegna vökva og salttaps um meltingarveg vegna niðurgangs. Natríum getur tapast um nýru, oft vegna notkunar þvag-ræsilyfja en einnig getur verið um salttapandi nýrnabólgu, skort á saltsterum eða heilkenni salttaps frá heila í kjölfar höfuðáverka að ræða.2 Óhófleg seyting á vasópressíni er mjög sjaldgæf orsök natríumskorts hjá börnum en slíkt ástand er aftur mun algengara hjá fullorðnum.1 Ónóg inntaka natríums er sjaldgæf orsök en of mikil inntaka á vatni eða vanþrýstnum (e. hypotonic) vökva er ekki óalgeng hjá börnum. Í vökvaþurrðarástandi, eins og oftast er raunin við natríumskort, minnkar gaukulsíunarhraði og útskilnaður á natríum í þvagi minnkar. Sé natríumtapið frá öðrum stöðum líkamans mælist natríum í þvagi einnig lágt sem einnig væri raunin fyrir sjúklinga með minnkað virkt blóðrúmmál, eins og sjá má til dæmis í hjartabilun eða nýrungaheilkenni (e. nephrotic syndrome). Ef salttapið er um nýru myndu natríumgildi í þvagi aukast og þvagmagn einnig. Frumkomið salttap um nýru er helst að sjá hjá sjúklingum með arfgengan fjölblöðrunýrnasjúkdóm, bráða millivefsnýrabólgu eða króníska nýrnabilun. Saltsteraskortur, pseudóhypóaldósterónismus (PHA), notkun þvagræsilyfja og kvillar í meltingarfærum geta einnig leitt til aukins salttaps um nýru og natríumskorts í kjölfarið.3
Stýring natríumbúskaps er annars vegar háð flæði natríumjóna yfir himnur nýrnapíplufruma og hins vegar háð hormónum eins og aldósteróni. Aldósterón hefur tveimur meginhlutverkum að gegna. Annars vegar að stýra magni utanfrumuvökva og hins vegar að stýra saltjafnvægi líkamans, þá einkum natríum og kalíum, en hefur einnig óbein áhrif á vetnisjónaseytingu í nýrum. Í nýrnafrumum örvar aldósterón jónaflutning með því að auka virkni og myndun bæði Na+/K+ ATPasa dælu og sérstakra opinna natríumganga (e. epithelial sodium channel, ENaC). Áhrifum aldósteróns er miðlað gegnum viðtaka þess, saltsteraviðtakann (MR). Kalíumofgnótt er einkennandi fyrir aldósterónskort eða tornæmi nýrnapípla fyrir hormóninu. Þegar virkni aldósteróns vantar verður minni seyting á kalíum og vetnisjónum í nýrnapíplum auk þess sem endurupptaka á natríum verður minni.2 Samfara kalíumofgnótt er alltaf hætta á lífshættulegum hjartsláttartruflunum, eins og var raunin í tilfelli drengsins. Þessi hætta er meiri sé blóðsýring til staðar.
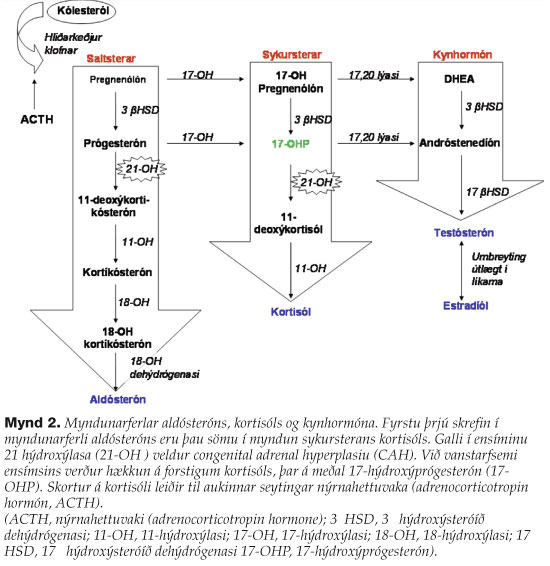
Í fyrstu var orsök salttruflana drengsins talin vera CAH sem er algengasta orsök aldósterónskorts hjá ungabörnum. Fjögur ensím hvata myndunarferli aldósteróns í zona glomerulosa í nýrnahettuberki. Fyrstu þrjú skrefin í ferlinu eru sömu skref og í myndun sykursterans kortisól-s í zona fasiculata en seinni skref í myndunarferlunum eru mismunandi. CAH er orsakað af víkjandi erfðagalla og er 21-hýdroxýlasa (21-OH, mynd 2) skortur langalgengastur. Ensímið hvatar skrefum í sameiginlegu myndunarferli kortisóls og aldósteróns og við vanstarfsemi ensímsins verður hækkun á forstigum kortisóls, þar á meðal 17-OHP. Skortur á kortisóli leiðir til aukinnar seytingar nýrnahettuvaka (adrenocorticotropin hormón, ACTH) (mynd 2). 17-OHP er veikur steri sem leiðir til karllegra einkenna á ytri kynfærum stúlkna en hefur lítil sem engin áhrif á útlit ytri kynfæra drengja. Drengir greinast því gjarnan seinna í ferlinu og er salttapandi krísa vegna aldósterónskorts oft fyrsta einkenni CAH. Ungbörn með salttapandi CAH fá vanalega einkenni á fyrstu tveimur vikum lífs og geta verið í lífshættu ef greining tefst. CAH var útilokað vegna lágs 17- OHP.4,5
{kind=link}
Sú brenglun blóðsalta sem drengurinn var með getur einnig orsakast af sjaldgæfum göllum svo sem PHA (galli er í saltsteraviðtakanum eða jónagöngum) og skorti á aldósterón synþetasa (ensím sem hvatar lokaskref aldósterónmyndunar). Aldósterón synþetasa skortur var útilokaður vegna aldósterónhækkunar.4,5 PHA var fyrst lýst 1958 af Cheek og Perry og hefur síðan þá verið flokkaður í tvo flokka sem innbyrðis eru ólíkir, klassíska formið eða PHA týpu 1 (PHA1) og PHA týpu 2 (PHA2). Utan við þessa flokkun hefur áunnum PHA einnig verið lýst sem er tímabundinn og afar sjaldgæfur. Áunnum PHA hefur verið lýst í ungbörnum með þvagrennslishindrun eða þvagfærasýkingu og lagast með öllu með meðferð undirliggjandi vanda.6,7PHA2 er einnig mjög sjaldgæfur sjúkdómur og kemur vanalega ekki fram nema í eldri börnum og fullorðnum. Sjúkdómurinn erfist með ríkjandi hætti og kemur til vegna galla í stýringu þíasíðnæmra Na+/Cl- ganga. PHA1 getur verið af tvennum toga, annars vegar sjúkdómur sem einskorðast við nýru og erfist með ríkjandi erfðamynstri (e. autosomal dominant PHA1, adPHA1) og hins vegar sjúkdómur sem leggst á fleiri líffæri líkamans og erfist víkjandi (e. autosomal recessive PHA1, arPHA1). Sjúklingar með arPHA1 þjást af lífshættulegu salttapi og kalíumofgnótt sem leiðir til mikilla veikinda á fyrstu vikum lífs og þarfnast þeir ævilangrar meðferðar með stórum skömmtum af natríumuppbót og kalíumbindandi resínum til að halda söltum í jafnvægi. Orsök arPHA1 eru stökkbreytingar í undireiningum ENaC jónaganganna en utan nýrna má einnig finna göngin í ristli, svitakirtlum, munnvatnskirtlum og lungnaþekju. Endurteknum öndunarfærakvillum hefur verið lýst í þessum börnum vegna ónógrar upptöku vökva frá öndunarfæraþekjunni sem rekja má til galla í ENaC jónagöngunum.6,8 AdPHA1 hefur svipaða klíníska mynd og arPHA1 en hefur vanalega mun mildari gang þar sem salttap er eingöngu bundið við nýru. Orsök adPHA1 er galli í geni sem kóðar fyrir MR í nýrum. Einnig eru til dæmi um nýjar stakstæðar stökkbreytingar í MR geninu sem leiða til adPHA1 án þess að um fjölskyldusögu sé að ræða. MR genið er að finna á litningi fjögur og hefur 22 mismunandi stökkbreytingum verið lýst, ýmist ríkjandi breytingar eða stakstæðar, en allar leiða þær til taps á virkni viðtakans.9 Ekki hafa fundist arfhreinar MR stökkbreytingar sem bendir til þess að algert tap á MR virkni samræmist ekki lífi. Börn með adPHA1 geta annaðhvort verið einkennalaus eða merki um marktækt salttap koma fram á fyrstu vikum lífs. Ekki er að fullu ljóst hvers vegna sum börn fá klínísk einkenni sjúkdómsins en önnur ekki en komið hafa fram hugmyndir um að einungis þegar álag er á stjórnun saltjafnvægis, eins og verður við ýmis veikindi eins og niðurgang eða þvagfærasýkingar, þá komi einkenni sjúkdómsins fram. Komi til salttaps þurfa börnin uppbótarmeðferðar við með natríum og stundum einnig kalíumbindandi resínum. Einkenni hverfa þó oftast þegar börnin eldast og ná þau þá að viðhalda saltjafnvægi án meðferðar með því að keyra upp renín-angiotensín-aldósterónkerfið og aðlaga mataræði að salttapinu 6,8,9.
Hjá drengnum sem hér um ræðir hafa ekki fundist þekktar stökkbreytingar í MR geninu og virðast foreldrar hans ekki vera með sama vandamál þar sem aldósteróngildi þeirra voru eðlileg. Í upphafi reyndist drengurinn því annaðhvort vera með áunna PHA vegna þvagfærasýkingar eða PHA1 sem hafði orsakast af óþekktri stakstæðri stökkbreytingu í MR geninu til dæmis brottfall eða endurröðun sem stundum greinast ekki með raðgreiningu. Þar sem gildi aldósteróns er nú innan eðlilegra marka má telja líklegra að um áunna PHA hafi verið að ræða. Hvað veldur tornæmi nýrnapípla fyrir aldósteróni er enn óþekkt og væri vert að rannsaka frekar. Menn greinir á um hvort bakteríusýkingunni sjálfri sé um að kenna eða galla í byggingu þvagkerfa sem margir þessara sjúklinga hafa. Sú staðreynd að margar mismunandi bakteríur hafa verið greindar í þessu ástandi og að meðferð við þvagfærasýkingunni veldur því að einkenni ganga algerlega til baka auk þess sem þeim er haldið niðri með fyrirbyggjandi sýklalyfjagjöf bendir frekar til þess að bólgan í nýrunum sjálfum ýti undir tornæmið.10 Eitt er þó víst að drengurinn sem um ræðir hafði klassísk einkenni PHA, natríumskort, kalíumofgnótt og blóðsýringu og minnir okkur á mikilvægi þess að athuga blóðsölt hjá ungum börnum með efri þvagfærasýkingu.
Heimildir
- Gonc EN, Kandemir N, Sen Y, Yordam N. Hyponatremia can be a presenting finding of multiple pituitary hormone deficiency in children: report of a case and review of literature. Clin Pediatr (Phila) 2005; 44: 623-8.
- Limal J-M. Hormonal Regulation of Sodium Metabolism. In: Bertrand J, Rappaport R, Sizonenko PC, eds. Pediatric endocrinology: physiology, pathophysiology & clinical aspects. 2 ed. Williams & Wilkins, Baltimore 1993: 544-54.
- Muglia LJ, Majzoub JA. Disorders of the Posterior Pituitary. In: Foley F, ed. Pediatric Endocrinology. Second Edition ed: Saunders, 2002: 289-322.
- White PC. Disorders of aldosterone biosynthesis and action. N Engl J Med 1994; 331: 250-8.
- White PC. Aldosterone synthase deficiency and related disorders. Mol Cell Endocrinol 2004; 217: 81-7.
- Geller DS. Mineralocorticoid resistance. Clin Endocrinol (Oxf) 2005; 62: 513-20.
- Geller DS, Rodriguez-Soriano J, Vallo Boado A, et al. Mutations in the mineralocorticoid receptor gene cause autosomal dominant pseudohypoaldosteronism type I. Nat Genet 1998; 19: 279-81.
- Zennaro MC, Lombes M. Mineralocorticoid resistance. Trends Endocrinol Metab 2004; 15: 264-70.
- Geller DS, Zhang J, Zennaro MC, et al. Autosomal dominant pseudohypoaldosteronism type 1: mechanisms, evidence for neonatal lethality, and phenotypic expression in adults. J Am Soc Nephrol 2006; 17: 1429-36.
- Nandagopal R, Vaidyanathan P, Kaplowitz P. Transient Pseudohypoaldosteronism due to Urinary Tract Infection in Infancy: A Report of 4 Cases. Int J Pediatr Endocrinol 2009;2009:195728(Epub 2009 May 21).