
05.tbl. 112. árg. 2026
Fræðigrein
Yfirlitsgrein. Huntington-sjúkdómur
doi 10.17992/lbl.2026.05.891
Fyrirspurnir sendist til Ólafs Sveinssonar, olafursv@landspitali.is
Greinin barst 19. janúar 2026, samþykkt til birtingar 7. apríl 2026
Ágrip
Inngangur
Huntington-sjúkdómur (HS) er sjaldgæfur, eingena, ríkjandi taugahrörnunarsjúkdómur sem einkennist af stigvaxandi hreyfitruflunum, geðrænum einkennum og vitrænni skerðingu. Sjúkdómurinn stafar af auknum fjölda endurtekninga á núkleótíðunum cýtósín-adenín-gúanín (CAG) í huntingtin-geninu sem leiðir til myndunar huntingtin-próteins með skaðlega virkni og uppsöfnunar próteinkekkja í taugafrumum sem veldur taugahrörnun. Algengi HS hefur áður mælst lágt á Íslandi samanborið við önnur Evrópulönd en nýlegar íslenskar rannsóknir benda til aukins algengis, líklega vegna bættrar greiningarhæfni og aukinnar notkunar erfðaprófa. Greining byggir á klínísku mati, fjölskyldusögu og staðfestingu með erfðaprófi. Engin sjúkdómsbreytandi meðferð er tiltæk í dag en einkennameðferð og þverfagleg nálgun geta bætt færni og lífsgæði sjúklinga. Í þessari yfirlitsgrein er fjallað um faraldsfræði, erfðafræði, klínísk einkenni, greiningu og meðferð Huntington-sjúkdóms, með sérstakri áherslu á nýlegar íslenskar rannsóknarniðurstöður og horfur til framtíðar.
Inngangur
Huntington-sjúkdómur (HS) er sjaldgæfur, ættlægur tauga-hrörn-unarsjúkdómur sem einkennist af ofhreyfingum (chorea), geðröskunum og vitrænni skerðingu. George Huntington lýsti sjúkdómnum árið 1872. Faðir hans og afi höfðu verið heimilis-læknar á sama svæði samfleytt í alls 78 ár og George fylgdi þeim í heimavitjanir. Þessi langi tími í eftirfylgd sjúklinga gaf honum einstakt sjónarhorn á sjúkdóminn. Huntington, þá 22 ára unglæknir í New York fylki í Bandaríkjunum, lýsti ýtarlega einkennum sjúkdómsins og erfanleika hans í greininni „On Chorea.“1 Í greininni útskýrði Huntington að ef börnin fara í gegnum lífið án þess að veikjast, slitnar þráðurinn og barnabörn geta verið viss um að þau séu laus undan sjúkdóminum. Einnig lýsti hann geðrænum veikindum sjúklinga og tilhneigingunni til sjálfsvígs og að í flestum tilvikum hæfist sjúkdómurinn ekki fyrr á fullorðinsárum.1
Faraldsfræði
HS fyrirfinnst alls staðar í heiminum en þó sérstaklega í ein-stak-lingum af evrópskum uppruna. Í nýlegri safgngreiningu (meta-analysis) reyndist áætlað algengi HD á heimsvísu vera 4,88 á hverja 100.000 íbúa og 6,37 í Evrópu2 en hefur mælst töluvert hærra í einstaka rannsóknum frá Kanada (13,7)3, Englandi (12,3)4 og Norður-Írlandi (10,6).5 Þessi aukning stafar líklega af innleiðingu erfðaprófa sem gera kleift að greina meinvaldandi breytingar í sjúklingum án einkenna og fjölskyldusögu, sem og aldraða sjúklinga með væg einkenni. Lengri lifun HS-sjúklinga eykur einnig algengið. Algengið í Japan, Tævan og Hong Kong er allt að tífalt lægra en í Evrópulöndum. Einstaklingar af evrópskum uppruna bera að meðaltali fleiri CAG endurtekningar í HTTgeninu en ein-staklingar af asískum eða afrískum uppruna.6
Hæstu algengistölur hafa sést á landfræðilega afmörkuðum svæðum þar sem ákveðnar fjölskyldur hafa einangrast í langan tíma. Besta dæmið er samfélag í kringum Maracaibo-vatnið í Venesúela. Þar hafa nokkur hundruð tilfella verið rakin til sama forföður og algengi mælst 700 á hverja 100.000 íbúa. Kerfisbundin rannsókn á þessu afmarkaða þýði hefur verið í gangi síðan árið 19817 og spilaði hún stórt hlutverk í einangrun meingensins.8 Auk þess er um að ræða klíníska langtímarannsókn á samfélagi án aðgangs að sértækri læknisþjónustu, sem gefur innsýn í náttúrulegan gang sjúkdómsins. Nokkuð er um arfhreina einstaklinga á þessu svæði og sýnt hefur verið fram á að þeir eru ekki verr settir af sjúkdómnum en arfblendnir.9
Í íslenskri rannsókn frá árinu 2007 mældist algengið hér á landi 0,96 tilfelli á hverja 100.000 íbúa.10 Til eru eldri innlendar algengistölur frá árinu 1961 og mældist algengi þá 2,67 tilfelli á hverja 100.000 íbúa sem er frekar lágt.11 Rannsóknin 2007 benti til þess að 31 einstaklingur hafi verið greindur með HS á Íslandi frá miðri 19. öld og að þeir voru allir afkomendur hjóna sem bjuggu á Norðurlandi.10 Í nýrri rannsókn sem náði yfir tímabilið 2008-2022 greindust 22 einstaklingar (ellefu karlar og ellefu konur). Fimm einstaklingar létust og voru þar með 17 á lífi við lok rannsóknartímabilsins og var algengið 4,38 tilfelli á hverja 100.000 íbúa. Algengi sjúkdómsins mældist því nokkuð hærra miðað við árið 2007. Ýmsar ástæður geta legið hér að baki, en helst ber að nefna betri greiningarhæfni. Aðeins tveir einstaklingar voru greindir með erfðarannsókn í fyrri rannsókninni en í þeirri nýju voru þeir 21 af 22. Einungis var þekkt ein stór íslensk HS-fjölskylda hér á landi 2007 en síðan hafa stök tilfelli komið fram, fleiri fjölskyldur bæst við ásamt því að erlendir einstaklingar með meinvaldandi breytinguna hafa flutt til landsins.12
Þrátt fyrir að algengi hafi hækkað nokkuð á Íslandi er það enn lægra en almennt í Evrópu, þar sem það er um 6,37 tilfelli á hverja 100.000 íbúa.2 Hið lága algengi á Íslandi kemur nokkuð á óvart í ljósi uppruna okkar frá Norðurlöndum, Írlandi og Skotlandi þar sem algengið hefur mælst töluvert hærra.4,13 Mögulegar skýringar á þessu eru landnemaáhrif (founder effect) og genaflökt (genetic drift), sem eiga sér stað fyrir tilviljun þegar stofn verður til út frá fáum landnemum í einangruðum samfélögum. Einnig hefur mögulega verið sterkara val gegn því að bera breytinguna á Íslandi, auk flutnings fólks úr landi, ekki síst frá Norðurlandi, til Nýja heimsins. Ólíklegt er að hið lága algengi skýrist af slæmri greiningarhæfni eða slælegri heilbrigðisþjónustu þar sem einkenni sjúkdómsins eru yfirleitt nokkuð sláandi. Finnland er einnig dæmi um erfðafræðilega einangrað land og er algengi sjúkdómsins þar einungis 2,12 tilfelli á hverja 100.000 íbúa.2
Erfðafræði
HS er eingena, ríkjandi erfðasjúkdómur. Það eru því helmingslíkur á að afkvæmi foreldris með HS erfi meinvaldandi breytinguna og fái sjúkdóminn, lifi það nógu lengi. Sjúkdómurinn er einn af mörgum sem tengjast aukningu á fjölda endurtekninga á þremur núkleótíðum (trinucleotide repeat expansion) í viðkomandi geni. Breytingin í HS er endurtekning á núkleótíðunum cýtidín-adenósín-guanósín (CAG) í fyrstu útröð HTT-gensins sem er staðsett á stutta armi litnings 4 (4p16.3) og tjáir fyrir huntingtin-próteini (HTT).14 Þar sem CAG táknar fyrir glútamín myndar N-endi HTT-próteinsins með meinvaldandi breytingu (mHTT) óeðlilega langa glútamínröð (polyglutamine tract). Eðlilegt HTT-gen er með ≤26 CAG-endurtekningar; millistærðarsamsæta ber 27-35 endurtekningar og veldur ekki Huntington-sjúkdómi en fjöldi endurtekninga getur verið óstöðugur. Ófullkomin sýnd, oft með hægfara sjúkdómsgang, kemur fram við 36-39 endurtekningar og fullkomin sýnd við ≥40 endurtekningar.15 Almennt gildir að því fleiri sem endurtekningarnar eru, því verri verður sjúkdómsmyndin og því fyrr koma einkenni fram.16 Algengast fyrir HS-sjúklinga er að hafa 40-45 endurtekningar og þá er nokkuð breytilegur upphafstími einkenna,17 sem skýrist meðal annars af erfðabreytileika í DNA-viðgerðarferlum (til dæmis MLH1) líkamsfruma18 og umhverfisþáttum.7 Til er barna- og unglingaform HS, þar sem einkenni koma fram fyrir tuttugu ára aldur og eru CAG endurtekningar þá >55.19 Sé einstaklingur með fjölda endurtekninga á bilinu 36-39 kemur til greina að rannsaka sérstaklega hvort hann sé með röðina CAA eða CAG í næstaftasta táknanum. Báðar framangreindar raðir eru tákn fyrir glútamín en CAA-kóðinn er algengari. Það sýnir sig að ein-staklingar með CAG-kóðann í þessari stöðu eru með meiri óstöðug-leika í fjölda endurtekinna raða í erfðaefni líkamsfrumna. Þeir fá frekar einkenni Huntington-sjúkdóms og fyrr á ævinni.20
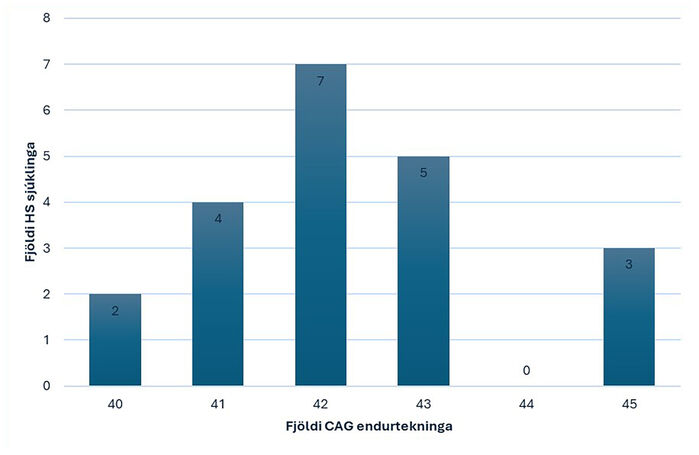
Mynd 1. Dreifing CAG endurtekninga meðal Huntington-sjúklinga á Íslandi.
Í nýlegri íslenskri rannsókn var meðalfjöldi CAG-endur-tekn-inga 42,3 (miðgildi 42).12 Hæsta gildi CAG-endurtekninga var 45 og lægsta gildi 40. Dreifingu endurtekninganna meðal HS-sjúklinga á Íslandi má sjá á mynd 1. Í þýðinu erfðu 13 sjúklingar (65%) sjúkdóminn frá feðrum sínum og sjö frá mæðrum (35%).12 Miðgildi CAG-endurtekninga í erlendum rannsóknum er í kringum 44, sem bendir til þess að fjöldi CAG-endurtekninga sé að jafnaði lægri meðal HS-sjúklinga á Íslandi. Á Íslandi voru 9,1% tilfellanna stök, án fjölskyldusögu. Þetta samræmist erlendum rannsóknum.21
CAG-endurtekningarnar eru óstöðugar í kynfrumum, sem þýðir að fjöldi þeirra hefur tilhneigingu til að breytast á milli kynslóða. Þó að fækkun geti átt sér stað er einkennandi að fjöldi endurtekninga eykst með hverri kynslóð. Þetta fyrirbæri kallast væntingarerfðir (anticipation) og það hefur tvennt í för með sér. Annars vegar að afkvæmi heilbrigðra einstaklinga með 27-35 endurtekningar geta fengið sjúkdóminn. Hins vegar að afkvæmi HS-sjúklinga geta veikst fyrr og fengið alvarlegri einkenni en foreldrið.22 Erfist breytingin frá móður er hún yfirleitt svipuð, með þrjár endurtekningar til eða frá. Erfist breytingin frá föður er sterkari tilhneiging til þess að fjöldi endurtekninga aukist við kynfrumumyndun. Það endurspeglast meðal annars í því að 70-90% þeirra með barna- og unglingaform HS erfa breytinguna frá föður.23 Skýringin á þessu er talin vera sívirkni sæðismyndunar sem skapar meiri óstöðugleika en eggfrumumyndun.24
Meinmyndun
HTT er mjög stórt, 350 kílódalton (kDa), umfrymisprótein sem er tjáð í mörgum vefjum en þó mest í heilanum. Hlutverk þess er ekki fullkomlega þekkt en það hlýtur að vera mikilvægt þar sem próteinið er bæði vel varðveitt í dýraríkinu og mýs með óvirkt HTT-gen deyja á fósturstigi.25 Einnig tengist það mörgum frumuferlum á borð við blöðruflutning, innfrumun, bifháramyndun, sjálfsát, frumuskiptingu og umritun. Sennilega sinnir HTT hlutverki sínu með því að vera eins konar grind fyrir myndun próteinflóka.26
N-endi huntingtin-próteinsins með meinvaldandi breytingu (mHTT), samanstendur af óeðlilega langri glútamínröð sem gegnir lykilhlutverki í meinmyndun sjúkdómsins.27 Talið er að það verði svokölluð eitruð nývirkni (toxic gain of function). Huntingtín-breytingin veldur því að gallaða próteinið hefur eituráhrif á nærliggjandi frumur. Frávik í mRNA-splæsingu leiðir til þess að brot úr fyrstu útröð HTT-gensins klippist frá, það verður próteinrof á mHTT og próteinbrotin safnast saman og mynda útfellingar (aggregates). Þrátt fyrir að hunt-ingtin-próteinið sé til staðar í mörgum vefjum líkamans virðast eituráhrif próteinsins nánast einungis vera bundin við miðtaugakerfið.27 Hægt er að sjá próteinútfellingar bæði í kjarna og umfrymi frumna taugunga. Áhrifin fela í sér víðtæka truflun á starfsemi umritunar, hvatbera, taugamóta og flutningi taugaboða.28
Við krufningu er heili sjúklinga almennt rýr en mestu áhrifin sjást í rákakjarna (corpus striatum) og þá sérstaklega í rófukjarna (n. caudatus). Einnig er hrörnun í berki heilans. Rákakjarni samanstendur fyrst og fremst af GABA-taugafrumum (median spiny neurons, MSN) og þær eru næmastar fyrir áhrifum mHTT.27 Fyrst tapast MSN sem tengjast hliðlægum bleikhnetti (globus pallidus) og það er talið orsaka ofhreyfingarnar. Við frekari versnun á sjúkdómnum tapast MSN sem tengjast miðlægum bleikhnetti og það er talið orsaka stífleikann og truflaða vöðvaspennu (dystonia) síðar í sjúkdóminum. Elliglöp sjúkdómsins eru talin stafa af skemmdum í berki heilans, í djúpum kjörnum og taugabrautum frá djúpum kjörnum fram í ennishluta heilans (frontostriatal circuit). Í langt gengnum tilfellum geta allar taugafrumurnar verið horfnar úr rákakjarna og taugaörvefur (gliosis) komið í staðinn. Þegar hér er komið eru ofhreyfingarnar horfnar en trufluð vöðvaspenna og stífleiki situr eftir (akinetic-rigid state). Viðtökum fyrir dópamíni, asetýlkólíni og N-metýl-D-aspart (NMDA) fækkar mjög í rákakjarna og berki heilans. Þess má þó geta að tilraunameðferð með GABA eða acetýlkólínörvum hefur ekki skilað tilætluðum árangri.27,29
Klínísk einkenni
Einkennin koma yfirleitt fram eftir barneignaraldur og því viðhelst sjúkdómurinn í ættinni. Venjulega er upphaf einkenna milli þrítugs og fertugs en þó getur sjúkdómurinn byrjað frá fimm ára aldri til áttræðis.30 5-10% sjúklinga eru með barna- og unglingaform sjúkdómsins og fá einkenni fyrir tvítugt.19 Í nýlegri íslenskri rannsókn var meðalaldur við upphaf einkenna 46,3 ár (19-63 ár) og meðalaldur við greiningu var 49,7 ár. Barna- og unglingaform sjúkdómsins þekkist ekki hér á landi. Meðalaldur við upphaf einkenna á heimsvísu er talinn vera um 40 ár. Upphaf einkenna hjá Huntington-sjúklingum hér á landi er því í hærri kantinum miðað við önnur þýði.12
Þrjú meginform einkenna sjúkdómsins eru hreyfitruflanir, geðræn einkenni og vitsmunaleg skerðing. Þau geta öll hafist á sama tíma en einnig geta liðið mörg ár á milli. Yfirleitt koma einkennin hægt fram og eru oft væg í byrjun. Dæmi um væg einkenni í upphafi eru klaufska, eirðarleysi, pirringur og vanhirða. Þessi einkenni þróast gjarnan yfir í ofhreyfingar, verri andlega líðan og að lokum heilabilun. Mikið þyngdartap er algengt en það orsakast af ofhreyfingu og kyngingartregðu. Barna- og unglingaformið einkennist frekar af parkinsonlíkum einkennum, hæghreyfingum, stífleika, skjálfta og flogum.19
Í íslensku rannsókninni voru ofhreyfingar algengasta einkennið (80%). Önnur algeng hreyfieinkenni voru jafnvægistruflanir (50%) og skertar fínhreyfingar (45%). Algengustu geðrænu einkennin á rannsóknartímabilinu voru þunglyndi (65%), kvíði (50%) og skapsveiflur (50%) en svefnvandamál, sjálfsvígshugsanir, ranghugmyndir og þráhyggja/árátta voru einnig algeng geðræn einkenni. Minnistap (60%) var algengasta vitræna einkennið.12 Einkenni Huntington-sjúklinga á Íslandi samræmdust því sem talið er vera algeng einkenni á heimsvísu.
Ofhreyfingar
Ofhreyfingar eru mest áberandi og best þekkta einkennið, enda er sjúkdómurinn þekktur undir nafninu Huntington´s chorea. Ofhreyfingar eru óviljastýrðar, skyndilegar, rykkjóttar og óreglu-legar hreyfingar sem virðast danskenndar. Þær leggjast á viljastýrða vöðvahópa á handahófskenndan hátt og geta flætt frá einum vöðvahópi til annars. Nær- sem fjærvöðvar og áslægir (axial) vöðvar eru undirlagðir. Snemma í sjúkdómsganginum sjást andlitsgrettur, hreyfingar á augabrúnum og enni, axlaypptingar og rykkjóttar hreyfingar útlima. Við frekari versnun verða hreyfingar handa og fóta ýktari sem orsakar danskenndar hreyfingar og rykkjandi göngulag sem er einkennandi fyrir HS. Ofhreyfingarnar aukast við andlegt álag en hverfa í svefni. Með frekari versnun verða einfaldar athafnir daglegs lífs, svo sem tal og kynging, erfiðar. Á endanum hverfa ofhreyfingar og stífleiki og trufluð vöðvaspenna tekur yfir. Á því stigi er sjúklingurinn yfirleitt orðinn ósjálfbjarga hjúkrunarsjúklingur.31
Geðræn einkenni
Geðrænu einkennin geta komið fram sem pirringur, hvatvís hegðun, geðlægð, kvíði, þunglyndi eða skyndileg æðisköst. Persónuleikabreytingar eru þau einkenni sem reynast sjúklingi og ættingjum oft erfiðust. Hjá sumum sjúklingum koma fram geðrofseinkenni. Þessir sjúklingar eru því stundum grunaðir um geðklofa og vistast á geðdeildum. Sjálfsvígstíðni er mun algengari hjá HS-sjúklingum en hjá almennu þýði. Rannsóknir gerðar á sjálfsvígstíðni HS-sjúklinga sýna tíðnina vera á bilinu 4-6% en tilraunir til sjálfsvígs eru mun algengari, sérstaklega snemma í sjúkdómsferlinu.32
Vitræn einkenni
Skert vitræn geta er óhjákvæmileg í Huntington-sjúkdómnum. Yfirleitt er um að ræða vefræna heilabilun með stigvaxandi versnun á minni, tapi á vitsmunalegri getu, auknum sljóleika, áhugaleysi og minnkaðri umhirðu. Hugsunarferli sjúklinga verður hægara og erfitt verður að halda athygli, skipuleggja og framkvæma athafnir. Oft eru sjúklingar með skert innsæi á eigin einkenni og tímatengd verkefni reynast sjúklingum sérstaklega erfið.31
Önnur einkenni
Svefntruflanir eru algengar.31 Versnun verður á snöggum augn-hreyfingum (saccadic) og jafnri eftirfylgni (smooth pursuit). Þyngdartap er einnig algengt einkenni HS. Tal- og kyngingartruflun er algeng. Í langt gegnum tilfellum geta sjúklingar þróað með sér málstol. Beinþynning og vöðvarýrnun eru algeng. Hjartabilun á sér stað í 30% tilfella.19
Rannsóknir og greining
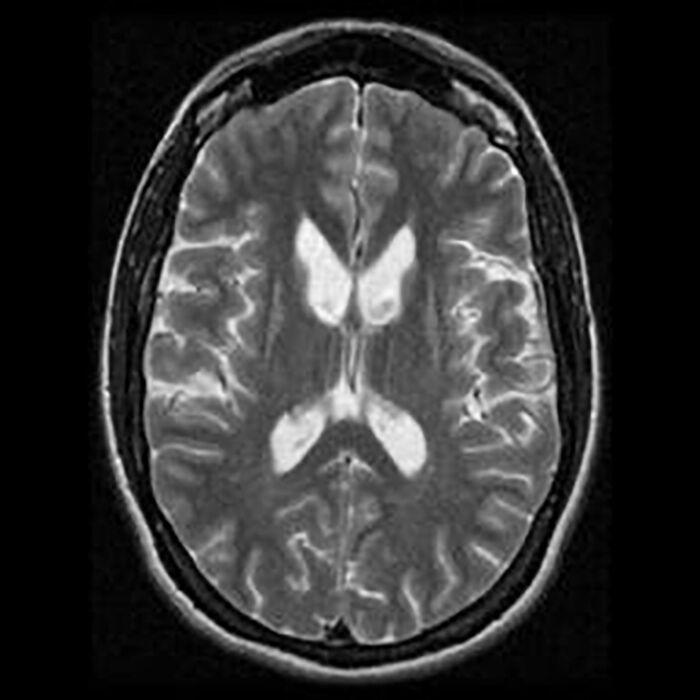
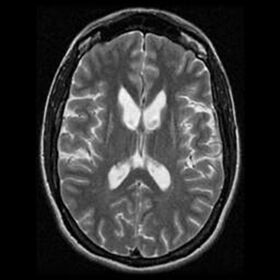
Mynd 2. Sýnir rýrnun á rófukjörnum sem liggja upp að hliðarhólfunum sem skapar „fiðrildisútlit“ vegna hrörnunar.
Almennar blóðrannsóknir eru eðlilegar. Myndrannsóknir á heila sýna stækkun á heilahólfum með einkennandi „fiðrildisútlit“ (mynd 2). Skapast það vegna hrörnunar á rófukjörnum sem liggja upp að hliðarhólfunum. Jáeindaskanni sýnir fram á minnkuð efnaskipti í rákakjarna.31
Greining HS byggir á fjölskyldusögu, taugaskoðun og erfðaprófi. Fyrir tíma erfðarannsókna var hægt að greina sjúk-dóm-inn með nokkurri vissu þegar saman komu ofhreyfingar, persónu-leikabreytingar, heilabilun og fjölskyldusaga. Dæmi um sjúkdóma sem geta líkst HS eru neuroacanthocytosis, Wilson-sjúkdómur, Sydenham-chorea, síðfettur (tardive dyskinesia), Tourette, Alzheimer-sjúkdómur og framheilabilun. Greiningin getur verið erfið ef engin fjölskyldusaga er til staðar og ber að hafa í huga að ekki þekkir fólk ávallt sína ættarsögu. Erfðapróf er nákvæmasta greiningartækið og örugg greining fæst með því að kanna fjölda CAG-endurtekninga.
Erfðaráðgjöf
Mikilvægt er að erfðaráðgjöf sé til staðar bæði fyrir og eftir greiningu. Á erfða- og sameindalæknisfræðideild Landspítala starfa erfðalæknar og erfðaráðgjafar. Hægt er að bóka tíma í gegnum Landspítala-appið án tilvísunar en fagaðilar geta sent tilvísun gegnum Heilsugátt. Leiðbeiningar eru aðgengilegar á síðu erfða- og sameindalæknisfræðideildar. Erfðaráðgjöf varðandi Huntington-sjúkdóm fylgir stöðluðu verklagi.
Forspárpróf
Fyrsta viðtal er við lækni og erfðaráðgjafa þar sem farið er yfir fjölskyldusögu og veittar upplýsingar um sjúkdóminn, erfðir, erfðamáta, áhættu, svartíma og framhald eftir að niðurstaða liggur fyrir. Sé einstaklingur ákveðinn í að fara í erfðarannsókn er boðið viðtal við sálfræðing nokkru eftir fyrsta viðtal. Þegar niðurstaða liggur fyrir, að jafnaði innan 8-12 vikna, er niðurstöðuviðtal. Í öll viðtöl er mælt með að einhver sé með viðkomandi, ráðþegi til stuðnings.
Fósturgreining og fósturvísagreining
Einstaklingar sem eru með aukinn fjölda endurtekninga í HTT-geni, eða eru með Huntington-sjúkdóm geta sótt um fósturvísagreiningu ef barneignir eru ráðgerðar.33,34 Erfða-ráðgjafar aðstoða við það ferli. Einnig er hægt að gera fósturgreiningu eftir 12 vikna meðgöngu.
Meðferð og sjúkdómsgangur
Engin meðferð er þekkt sem hægir á gangi sjúkdómsins en hægt er að beita einkennameðferð. Þverfagleg nálgun með teymi sem tekur til taugalækna, geðlækna, heimilislækna, sjúkraþjálfara, iðjuþjálfara, tal- og málþjálfa, næringarfræðinga og hjúkrunarfræðinga er afar æskileg.19
Við depurð og kvíða eru notuð geðdeyfðarlyf á borð við serótónín-endurupptökuhemla sem eru líklega van-nýtt meðferðarúrræði. Anddópamínvirk lyf eru notuð gegn óviljastýrðum hreyfingum en notkun þeirra takmarkast af stífleika og trufluðum vöðvasamdrætti. Mónóamínendurupptökuhemill-inn tetrabenazín er notaður gegn ofhreyfingum en forðast skal notkun lyfsins ef sterk saga er um þunglyndi eða sjálfsvígshugsanir vegna hættu á versnun þunglyndiseinkenna. Lyfið hindrar endurupptöku og losun dópamíns við taugamót í miðtaugakerfinu. Sýnt hefur verið fram á að lyfið leiðir til marktækrar minnkunar á ofhreyfingum samanborið við lyfleysu. Deutetrabenazín er skylt tetrabenazíni en hefur lengri helmingunartíma. Það er stundum valið umfram tetrabenazín þar sem skammtar eru færri og minni hætta er á þunglyndi.35,36 Í dag eru geðrofslyf á borð við olanzapín, risperidón, og halóperdól helst notuð en þau minnka áhrif dópamíns í miðtaugakerfinu ásamt því að hafa áhrif á geðræn einkenni sjúkdómsins. Þó ber að nefna að ofhreyfingarnar eru ekki ávallt truflandi fyrir sjúklinginn sjálfan. Geðrofslyf á borð við olanzapín eru notuð ef um geðrofslík einkenni er að ræða. Olanzapín hafði einnig áhrif á þunglyndi, kvíða, pirring og þráhyggju í rannsóknum.37 Valpróinsýra hefur einnig verið notuð við pirringi og skapsveiflum. Fyrir svefnvandamál er oft notað melatónín eða zópíklón.19
Í íslensku rannsókninni kom í ljós að algengasti lyfja-flokk-urinn sem HS-sjúklingar fengu var geðdeyfðarlyf, þar sem 85% fengu þau á rannsóknartímabilinu, 55% einstaklingar voru á geðrofslyfjum sem oft voru notuð fyrir ofhreyfingar, 45% á róandi lyfjum en einungis 5% á sértæku lyfi við ofhreyfingum. Um þriðjungur fékk svefnlyf á rannsóknartímabilinu.12
Sjúkraþjálfun og talþjálfun eru nauðsynlegar til að viðhalda sjálfstæði eins lengi og hægt er. Talþjálfun getur hjálpað með tjáningu. Talmeinafræðingur getur einnig gefið ráð hvað kyngingu varðar og viðeigandi mataræði. Einstaklingur með HS þarf næringarmikla fæðu því orkubrennsla er meiri en hefðbundið er vegna ofhreyfinga. Því getur verið æskilegt að leita til næringarráðgjafa og fylgjast þarf vel með þyngdartapi. Iðjuþjálfi getur hjálpað með athafnir daglegs lífs. Ýmis hjálpartæki og breyting á heimili til að minnka byltuhættu geta komið að góðum notum. Endanleg vistun á hjúkrunarstofnun á sér yfirleitt stað þegar sjúkdómurinn er langt genginn.19,38
Skoðun á sjúkragögnum Huntington-sjúklinga benti til þess að flestir sjúklingar hér á landi væru í virku eftirliti og fengju nokkuð góða einkennameðferð eins og tíðni lyfjameðferðar ber vitni um. Algengasta dánarorsök var ásvelgingslungnabólga, sem er í samræmi við aðrar rannsóknir á HS. Enginn einstak-lingur lést af völdum sjálfsvígs.12 Meðalaldur við andlát var 70,4 ár en í rannsókninni frá 2007 61,9 ár.10 Meðalaldurinn hefur hækkað talsvert hér á landi en það kann að skýrast af betri úrræðum fyrir HS-sjúklinga nú en fyrir 15 árum síðan. Í norskri rannsókn frá árinu 2018 var meðalaldur þeirra sem létust 63,9 ár og á heimsvísu 58 ár. Aldur Huntington-sjúklinga á Íslandi er því hærri, sem kann að skýrast af almennt lægri fjölda CAG-endurtekninga hér á landi.39 Meðalævilengd Íslendinga 2011-2020 var 82,7 ár samkvæmt Hagstofu Íslands. Lífslíkur HS- sjúklinga eru því skertar miðað við almennt þýði.
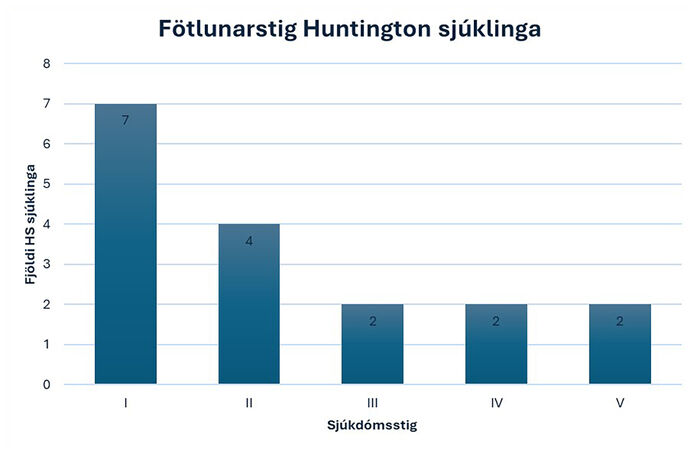
Mynd 3. Sýnir dreifingu fötlunarstigs sjúk-linga með Huntington-sjúkdóm á Íslandi sam-kvæmt Shoulson Fahn-stigunarkerfinu. Stig I-II lýsa snemmkomnum sjúkdómi þar sem sjúklingar eru alveg eða að mestu sjálfstæðir. Stig III lýsir miðlungs fötlun með talsverða skerðingu á daglegri færni en sjúklingar viðhalda að einhverju leyti sjálfstæði. Stig IV-V lýsa hins vegar langt gengnum sjúkdómi, með mikilli fötlun.
Í nýju íslensku rannsókninni var fötlunarstig sjúklinga með Huntington-sjúkdóm metið við lok rannsóknartímabilsins. Notað var Shoulson Fahn-stigunarkerfið sem byggir á klínísku mati á fötlun HS-sjúklinga út frá heildarfærni þeirra. Tekið er mið af þáttum eins og vinnufærni, fjármálastjórnun og athöfnum daglegs lífs. Mynd 3 sýnir fötlunarstig sjúklinganna samkvæmt flokkunarkerfi Shoulson Fahn í lok árs 2022. Dreifingin var tiltölulega jöfn en þó voru heldur fleiri á fyrstu stigum og því stutt komnir á sjúkdómsferlinu.12
Lyfjaþróun
Þrátt fyrir aukinn skilning á meinmyndun HS hefur árangur klínískra lyfjarannsókna verið býsna dræmur. Í samantekt á klínískum lyfjarannsóknum sem gerðar voru frá byrjun til ársins 2017 voru borin kennsl á 99 rannsóknir.40 Af þeim náðu aðeins 20 í fasa þrjú og aðeins hafa tvö lyf (tetrabenazín og deutetra-benazín, sjá að ofan) verið samþykkt en hvorugt þeirra hægir á gangi sjúkdómsins. Mest rannsökuðu efnasamböndin hafa verið kreatín, latrepírdín og prídópídín. Í dag er verið að rannsaka ýmis ný lyf og meðal þeirra ber hæst að nefna næstu kynslóðar meðferðir (next generation genetic therapies). Einnig eru nokkrar væntingar bundnar við stofnfrumumeðferðir.
Marksæknar RNA-meðferðir (RNA-targeted therapy) eru lengra komnar en marksæknar DNA-meðferðir. Mest rannsökuðu aðferðirnar eru andþætt ólígonúkleótíð (antisense oligonucleotides (ASOs, tjáningarhindrar) og RNA íhlutun (RNA interference (RNAi). Þær miða að því að minnka magn HTT mRNA og þar með huntingtin-próteinsins. Tveir taugahrörnunarsjúkdómar, Duchenne vöðvarýrnun (Duchenne muscular dystrophy) og mænutaugahrörnun (spinal muscular atrophy), eru með samþykkt ASO-lyf.
ASOs eru einþátta DNA-bútar sem eru mótsvarandi (com-plement-ary) við mHTT-mRNA. ASO bindast mHTT-mRNA sem leiðir til niðurbrots með hjálp RNAsaH og kemur þannig í veg fyrir myndun skaðlega próteinsins. Rannsóknir á HS-músamódelum hafa sýnt að ASO-tæknin minnkar bæði magn mHTT-mRNA og mHTT í heila og dregur úr HS-líkum einkennum í músunum.41 Birtar hafa verið niðurstöður úr slembiraðaðri og tvíblindri fasa 2a klínískri lyfjarannsókn á HS-sjúklingum með vægan sjúkdóm þar sem ASO-tæknin var notuð.42 Í rannsókninni var ASO gegn mHTT gefið í mænuvökva (intrathecal) í hækkandi skömmtum á fjögurra vikna fresti. Til að mæla áhrif meðferðarinnar var fylgst með breytingum á magni mHTT í mænuvökva. Niðurstöðurnar sýndu skammtaháða lækkun á mHTT í mænuvökva og hæsti skammturinn minnkaði mHTT um 38%. Ekki mældust neinar marktækar aukaverkanir hjá þátttakendum. Fasa 3 klínísk lyfjarannsókn reyndist þó neikvæð hvað varðar áhrif á klínísk einkenni sjúkdómsins.43
RNAi er hluti af náttúrulegu stjórnkerfi gena í heilkjörnungum og leiðir til bælingar á virkni gena. Hægt er að nota RNAi í meðferðarskyni til að lækka magn mHTT-mRNA með sér-hönnuðum RNA-brotum, svo sem small interfering RNA, (siRNA), short hairpin RNA (shRNA) eða microRNA (miRNA).44 RNA-brotin bindast mHTT mRNA gegnum RISC-flóka (RNA-induced silencing complex) og veldur niðurbroti þess. Vegna -blóð--heila-þröskuldsins þarf að gefa RNAi-meðferð í heilavef, oftast með veirugenaferjum. Þetta er ólíkt ASO sem hafa verið gefin í mænuvökva. Aftur á móti þarf að gefa veirugenaferjur mun sjaldnar þar sem áhrif vara lengur. Algengast er að nota adeno-associated veirugenaferjur (AAV) þar sem þær valda litlu ónæmissvari og innlimun þeirra í erfðaefni frumunnar er mjög lítil.44,45
Nýlega komu jákvæðar fréttir fyrir annars stigs fasa rannsókn Uniqure með AMT-130 (miRNA) sem gefið er inn í djúphnoð (bas-al ganglia) með segulómunarstýrðri taugaskurðaðgerð (óbirtar niðurstöður). Náði rannsóknin aðalmarkmiði sínu og sýndi í fyrsta skipti að lyf getur hægt á klínískum framgangi sjúk-dómsins. Notuð var adenotengd veirugenaferja (AAV).45
Samantekt
Huntington-sjúkdómur er sjaldgæfur, eingena, ríkjandi taugahrörnunarsjúkdómur sem einkennist af stigvaxandi hreyfitruflunum, geðrænum einkennum og vitrænni skerðingu. Sjúkdómurinn stafar af auknum fjölda endurtekninga á núkleótíðunum cýtósín-adenín-gúanín (CAG) í huntingtin-geninu (HTT) sem leiðir til myndunar huntingtin-próteins með skaðlega virkni og uppsöfnunar próteinbrota í taugafrumum sem veldur taugahrörnun. Algengi sjúkdómsins hefur áður mælst lágt á Íslandi samanborið við önnur Evrópulönd en nýlegar íslenskar rannsóknir benda til hærra algengis, líklega vegna bættrar greiningarhæfni og aukinnar notkunar erfðaprófa. Engin meðferð hefur fundist sem hægir á framgangi sjúkdómsins en nýlegar rannsóknir með veirugenaferju gefa vonir um árangur.
Heimildir
1. Harper PS. Huntington's disease in a historical context. In: Bates G, Tabrizi S, Jones L, editors. Huntington's disease. Oxford: Oxford University Press; 2014. p. 3–21.
2. Medina A, Correa M, Arango-Lasprilla JC, et al. Prevalence and incidence of Huntington's disease: an updated systematic review and meta-analysis. Mov Disord. 2022;37(12):2327–2335.
3. Fisher ER, Hayden MR. Multisource ascertainment of Huntington disease in Canada: prevalence and population at risk. Mov Disord. 2014;29(1):105–114.
4. Evans SJ, Douglas I, Rawlins MD, et al. Prevalence of adult Huntington's disease in the UK based on diagnoses recorded in general practice records. J Neurol Neurosurg Psychiatry. 2013;84(10):1156–1160.
5. Morrison PJ, Harding-Lester S, Bradley A. Uptake of Huntington disease predictive testing in a complete population. Clin Genet. 2011;80:281–286.
6. Bates GP, Dorsey R, Gusella JF, et al. Huntington disease. Nat Rev Dis Primers. 2015;1:15005.
7. Wexler NS, Lorimer J, Porter J, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci U S A. 2004;101(10):3498–3503.
8. Gusella JF, Wexler NS, Conneally PM, et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature. 1983;306(5940):234–238.
9. Wexler NS, Young AB, Tanzi RE, et al. Homozygotes for Huntington's disease. Nature. 1987;326(6109):194–197.
10. Sveinsson O, Halldórsson S, Olafsson E. An unusually low prevalence of Huntington's disease in Iceland. Eur Neurol. 2012;68(1):48–51.
11. Gudmundsson KR. Prevalence and occurrence of some rare neurological diseases in Iceland. Acta Neurol Scand. 1969;45(1):114–118.
12. Briem S, Stefansdottir V, Jónsson JJ, et al. The epidemiology of Huntington's disease in Iceland. Eur Neurol. 2025;88(2):64–70.
13. Gilling M, Durr A, Abe K, et al. The Danish HD Registry: a nationwide family registry of HD families in Denmark. Clin Genet. 2017;92(3):338–341.
14. Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72(6):971–983.
15. Rubinsztein DC, Leggo J, Coles R, et al. Phenotypic characterization of individuals with 30–40 CAG repeats in the Huntington disease gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am J Hum Genet. 1996;59(1):16–22.
16. Andrew SE, Goldberg YP, Kremer B, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genet. 1993;4(4):398–403.
17. Duyao M, Ambrose C, Myers R, et al. Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat Genet. 1993;4(4):387–392.
18. Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington's disease. Cell. 2015;162(3):516–526.
19. Ghosh R, Tabrizi SJ. Huntington disease. Handb Clin Neurol. 2018;147:255–278.
20. Wright GEB, Collins JA, Kay C, et al. Length of uninterrupted CAG, independent of polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am J Hum Genet. 2019;104(6):1116–1126.
21. Warby SC, Montpetit A, Hayden AR, et al. CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am J Hum Genet. 2009;84(3):351–366.
22. Semaka A, Kay C, Doty CN, et al. CAG size-specific risk estimates for intermediate allele repeat instability in Huntington disease. J Med Genet. 2013;50(10):696–703.
23. Telenius H, Kremer B, Goldberg YP, et al. Molecular analysis of juvenile Huntington disease: the major influence on (CAG)n repeat length is the sex of the affected parent. Hum Mol Genet. 1993;2(10):1535–1540.
24. Kremer B, Goldberg YP, Andrew SE, et al. Sex-dependent mechanisms for expansions and contractions of the CAG repeat on affected Huntington disease chromosomes. Am J Hum Genet. 1995;57(2):343–350.
25. Nasir J, Floresco SB, O'Kusky JR, et al. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81(5):811–823.
26. Saudou F, Humbert S. The biology of huntingtin. Neuron. 2016;89(5):910–926.
27. McColgan P, Tabrizi SJ. Huntington's disease: a clinical review. Eur J Neurol. 2018;25(1):24–34.
28. Carroll JB, Bates GP, Steffan J, et al. Treating the whole body in Huntington's disease. Lancet Neurol. 2015;14(11):1135–1142.
29. Bonelli RM, Wenning GK, Kapfhammer HP. Huntington's disease: present treatments and future therapeutic modalities. Int Clin Psychopharmacol. 2004;19(2):51–62.
30. Brinkman RR, Mezei MM, Theilmann J, et al. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet. 1997;60(5):1202–1210.
31. Roos RAC. Clinical neurology. In: Bates G, Tabrizi S, Jones L, editors. Huntington's disease. Oxford: Oxford University Press; 2014. p. 123–145.
32. Grimaldi A, Veneziani I, Culicetto L, Quartarone A, Lo Buono V. Risk Factors and Interventions for Suicide in Huntington's Disease-A Systematic Review. J Clin Med. 2024 Jun 12;13(12):3437.
33. Roos RAC. Huntington's disease: a clinical review. Orphanet J Rare Dis. 2010;5:40.
34. MacLeod R, Tibben A. Genetic counseling and testing. In: Bates G, Tabrizi S, Jones L, editors. Huntington's disease. Oxford: Oxford University Press; 2014. p. 165–178.
35. Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology. 2006;66(3):366–372.
36. Guay DR. Tetrabenazine, a monoamine-depleting drug used in the treatment of hyperkinetic movement disorders. Am J Geriatr Pharmacother. 2010;8(4):331–373.
37. Paleacu D, Anca M, Giladi N. Olanzapine in Huntington's disease. Acta Neurol Scand. 2002;105(6):441–444.
38. Nance MA. Comprehensive care. In: Bates G, Tabrizi S, Jones L, editors. Huntington's disease. Oxford: Oxford University Press; 2014. p. 393–404.
39. Rodrigues FB, Abreu D, Damasio J, et al. Survival, mortality, causes and places of death in a European Huntington's disease prospective cohort. Mov Disord Clin Pract. 2017;4(5):737–742.
40. Travessa AM, Rodrigues FB, Mestre TA, et al. Fifteen years of clinical trials in Huntington's disease: a very low clinical drug development success rate. J Huntingtons Dis. 2017;6(2):157–163.
41. Kordasiewicz HB, Stanek LM, Wancewicz EV, et al. Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron. 2012;74(6):1031–1044.
42. Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, et al. Targeting huntingtin expression in patients with Huntington's disease. N Engl J Med. 2019;380(24):2307–2316.
43. Rook ME, Southwell AL. Antisense oligonucleotide therapy in Huntington disease. BioDrugs. 2022;36(2):105–119.
44. Shannon KM. Recent advances in the treatment of Huntington's disease. CNS Drugs. 2020;34(3):219–228.
45. Aguiar S, van der Gaag B, Cortese FAB. RNAi mechanisms in Huntington's disease therapy: siRNA versus shRNA. Transl Neurodegener. 2017;6(1):30.