
Arfgengur skortur á storkuþætti VII í íslenskri fjölskyldu
Ágrip
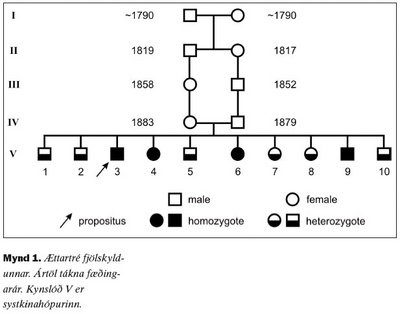
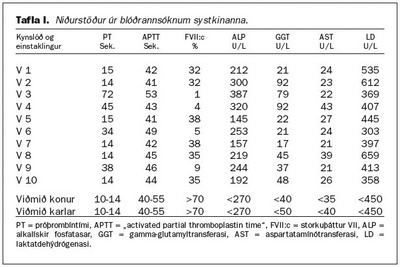
Lýst er sjaldgæfum blæðingasjúkdómi, ættgengum skorti á storkuþætti VII, í íslenskri fjölskyldu. Sjúkdómurinn erfist ókynbundið og yfirleitt víkjandi þannig að einkenni koma sjaldan fram nema þegar báðir erfðastofnarnir sem tjá storkuþátt VII eru gallaðir og mikill skortur er á storkuþætti VII. Tíu systkini reyndust öll hafa skort á storkuþætti VII. Fjögur höfðu skort á háu stigi og voru þess vegna sennilega með galla í báðum erfðastofnunum. Sex höfðu miðlungsskort og voru því sennilega með annan erfðastofninn eðlilegan en hinn gallaðan. Þetta bendir til þess að annað foreldri þeirra hafi haft galla í báðum erfðastofnum sínum en hitt aðeins í öðrum. Foreldrar þeirra voru þremenningar að skyldleika. Aðeins eitt af systkinunum hafði staðfesta blæðingahneigð. Það var karlmaður sem fékk endurteknar liðblæðingar frá barnsaldri og varð að lokum óvinnufær vegna liðskemmda. Ekkert af hinum systkinunum hafði greinileg merki um blæðingahneigð og engar sagnir voru um blæðingahneigð hjá forfeðrum eða formæðrum þeirra. Höfundum er ekki kunnugt um að þessi sjúkdómur hafi fundist hjá öðrum Íslendingum.
English Summary |
| Þorsteinsson V, Magnússon S, Hellman-Erlingsson S, Guðmundsdóttir BR, Árnason A Congenital deficiency of coagulation factor VII in an Icelandic family Læknablaðið 2004; 90: 385-8 Congenital deficiency of coagulation factor VII is a rare autosomal and usually recessive genetic bleeding disorder which has been discovered in an Icelandic family. The propositus is a male who experienced intermittent painful inflammation of his ankle joints at the age of 9-10 and later also in his knees, elbow, shoulder, and wrist. Smaller joints were spared, serologies for rheumatoid disease were negative. He was treated for rheumatoid arthritis with limited results and became practically invalid due to his arthritis at the age of 40. At the age of 57, a surgical synovectomy of his knee joint was complicated by postoperative bleeding, and signs of chronic haemorrhagic arthritis were noted in the synovia. Subsequently, a marked prolongation of his prothrombin time and a near total deficiency of coagulation factor VII were discovered. All of his nine siblings were deficient in coagulation factor VII, three of them markedly deficient like the proband and six moderetely deficient. The pattern of inheritance suggests that one of their parents was heterozygous and the other homozygous or doubly heterozygous of genetic deficiency of coagulation factor VII. The parents were second cousins. Of the siblings, only the propositus had a bleeding tendency or arthritis. No evidence of such symptoms in their parents or grandparents was found. This family is the only Icelandic family with congenital deficiency of coagulation factor VII known to the authors. Key words: coagulation factor VII, haemorrhagic disease. Correspondance: Vigfús Þorsteinsson, vigfus@fsa.is |
Inngangur
Sjúkratilfelli
Aðferðir
Niðurstöður
Umræða
Erfðavísirinn fyrir storkuþátt VII hefur verið kortlagður. Hann er á litningi 13q34 og er 12,8 kb að stærð. Storkuþátturinn sjálfur er glykóprótín með sameindarþunga um 50 kDa, framleitt í lifur með hjálp K-vítamíns. Storkuþátturinn hefur verið einangraður og bygging hans ákvörðuð. Hún er að mörgu leyti hliðstæð byggingu sumra annarra efna sem eru háð K-vítamíni í framleiðslu, til dæmis storkuþáttanna IX og X og prótíns C. Í blóðrásinni er storkuþáttur VII næstum allur í óvirku formi og án ensímvirkni. Hann verður ekki virkur við storkumyndun fyrr en hann hefur bæði örvast til ensímvirkni og bundist við vefjaþátt (tissue factor). Vefjaþáttur er prótín sem finnst á yfirborði flestra fruma nema blóðkorna og æðaþelsfruma, það er að segja þeirra frumna sem jafnan eru í snertingu við blóðið. Storkuþáttur VII og vefjaþáttur geta þannig aðeins náð saman ef blóðið kemst í snertingu við vefi utan blóðrásarinnar við rof á æð eða ef vefjaþáttur kemst inn í blóðrásina. Virkir storkuþættir, IXa, Xa, VIIa, XIIa (ensímvirkni er táknuð með -a) eða þrombín (IIa), örva storkuþátt VII til ensímvirkni með því að kljúfa hann. Hann getur þá tekið þátt í storkumyndun ef hann er bundinn við vefjaþátt en annars ekki. Samstæðan af virkum storkuþætti VII og vefjaþætti örvar storkuþætti IX og X til ensímvirkni. Sú örvun leiðir til myndunar próþrombínasa á yfirborði blóðflaga eða annarra frumna og próþrombínasi breytir síðan próþrombíni í þrombín. Þrombín er mikilvirkt ensím og lykilefni bæði í storkumyndun og storkuhindrun. Þrombín myndar síðan storku með því að breyta uppleystu fíbrínógeni í fast fíbrín (17).
Það virðist rökrétt að skortur á storkuþætti VII geti valdið blæðingum en að hann geti einnig valdið blóðtappahneigð, eins og áður var vikið að, virðist mótsagnakennt. Það hefur verið skýrt á þann hátt að örlítið af vefjaþætti sé stundum í snertingu við blóðið, til dæmis á yfirborði örvaðra stórkirninga (monocytes) eða þar sem æðaþel hefur skemmst. Storkuþáttur VII binst þá strax við vefjaþáttinn en örvast þó ekki til ensímvirkni ef virka storkuþætti vantar til að örva hann. Ef virkir storkuþættir, til dæmis IXa eða XIIa, myndast af einhverjum ástæðum í blóðrásinni bindast þeir strax við storkuþátt VII og örva hann, hvort sem hann er bundinn við vefjaþátt eða ekki. Hjá sjúklingum með skort á storkuþætti VII er hlutfallslega stærri hluti af storkuþættinum bundinn við vefjaþátt samanborið við heilbrigða þar sem stærsti hluti hans er óbundinn í blóðrásinni. Við skort á storkuþætti VII eru þannig meiri líkur á því að storkuþáttur IXa bindist við storkuþátt VII sem er þegar bundinn við vefjaþátt og þannig leiðir þessi ferill fremur til storkumyndunar hjá þeim sem hafa skort á storkuþætti VII en hjá hinum sem hafa ekki skort (18).
Á síðari árum hefur áhugi á storkuþætti VII farið vaxandi af ýmsum ástæðum. Ljóst er að hann gegnir lykilhlutverki við stöðvun blæðinga en kenningar um að hann gegni einnig stóru hlutverki í tilurð slagæðablóðtappa hafa vakið mikla athygli. Í Northwick Park Heart Study (19-21) og fleiri könnunum (22-26) kom fram jákvæð fylgni milli magns af storkuþætti VII í blóði og kransæðastíflu en engin slík tengsl hafa hins vegar fundist í öðrum sambærilegum könnunum (27-31). Hugsanlega tengist þetta misræmi í niðurstöðum því að mismunandi aðferðir við mælingar á virkni storkuþáttar VII hafa mismikið næmi fyrir mismunandi gerðum af storkuþættinum (32). Það ýtti einnig undir áhugann að tiltekin arfgerð af storkuþætti VII, Arg353---!Gln, þar sem amínósýran glútamín tekur sæti argíníns í 353. sæti í prótíninu, og jafnvel fleiri arfgerðir af storkuþættinum (33) hafa í för með sér minna magn af storkuþætti VII í blóði og samkvæmt sumum heimildum minni hættu á kransæðastíflu (34-36). Aðrar kannanir (37, 38) hafa þó ekki staðfest þessi tengsl þannig að enn er ekki ljóst hvern þátt mismunandi arfgerðir af storkuþætti VII eiga í tilurð kransæðastíflu. Ennfremur hafa tengsl storkuþáttar VII við þríglyseríð í blóði (39), fituneyslu (40) og fleiri þekkta áhættuþætti fyrir æðakölkun vakið athygli. Þá hefur storkuþáttur VII líka komist í sviðsljósið vegna framleiðslu á honum með erfðatækni og vaxandi notkunar við að stöðva blæðingar í ýmsum erfiðum blæðingasjúkdómum, til dæmis dreyrasýki A, þegar sjúklingar mynda mótefni gegn storkuþætti VIII sem torveldar mjög hefðbundna meðferð (41).
Þakkir
Heimildir
2. Owren PA, Aas K. The control of dicumarol therapy and the quantitative determination of prothrombin and proconvertin. Scand J Clin Lab Invest 1951; 3: 201-8.
3. Koller F, Loeliger A, Duckert F. Experiments on a new Clotting Factor (Factor VII). Acta Haematol 1951; 6: 1-18.
4. Owren PA. Proconvertin, the new clotting factor. Scand J Clin Lab Invest 1951; 3: 168.
5. Alexander B, Goldstein R, Landwehr G, Cook CD. Congenital SPCA deficiency: A hitherto unrecognized coagulation defect with hemorrhage rectified by serum and serum fractions. J Clin Invest 1951; 30: 596-608.
6. Peyvandi F, Mannucci PM. Rare Coagulation Disorders. Thromb Haemost 1999; 82: 1207-14.
7. Triplett DA, Brandt JT, McGann Batard MA, Schaeffer Dixon JL, Fair DS. Hereditary Factor VII Deficiency: Heterogeneity Defined by Combined Functional and Immunochemical Analysis. Blood 1985; 66: 1284-7.
8. Giansily-Blaizot M, Biron-Andreani C, Aguilar-Martinez P, de Moeloose P, Briquel M-E, Goudemand J, et al. Inherited factor VII deficiency and surgery: clinical data are the best criteria to predict the risk of bleeding. Br J Haematol 2002; 117: 172-5.
9. FVII mutation database at http://europium.csc.mrc.ac.uk
10. McMillan CW, Roberts HR. Congenital combined deficiency of coagulation factors II, VII, IX and X. Report of a case. N Engl J Med 1966; 274: 1313-5.
11. Girolami A, Sartori MT, Zerbinati P. Frequent association of factor VII defects with other clotting disorders. Blood Coag Fibrinol 1992; 3: 829-30.
12. Ly B, Solum NO, Vennerød AM, Dahl O, Hagen I, Ørstavik KH. A Syndrome of Factor VII Deficiency and Abnormal Platelet Release Reaction. Scand J Haematol 1978; 21: 206-14.
13. Seligsohn U, Shani M, Ramot B, Adam A, Sheba C. Dubin-Johnson Syndrome in Isarel. II Association with Factor-VII Deficiency. Q J Med 1970; 39: 569-84.
14. Godal HC, Madsen K, Nissen-Meyer R. Thrombo-Embolism in Patients with Total Proconvertin (Factor VII) Deficiency. A report on two Cases. Acta Med Scand 1962; 171: 325-7.
15. Gershwin ME, Gude JK. Deep Vein Thrombosis and Pulmonary Embolism in Congenital Factor VII Deficiency. N Engl J Med 1973; 288: 141-2.
16. Lefrere J-J, Chaunu M-P, Conrad J, Horellou M-H, Samana M. Congenital factor VII deficiency and cerebrovascular stroke. Lancet, 1985; 2: 1006-7.
17. Dahlbäck B. Blood coagulation. Lancet 2000; 355; 1627-32.
18. Østerud B. Factor VII and haemostasis. Blood Coag Fibrinol 1990; 1: 175-82.
19. Meade TW, Mellows S, Brozovic M, Miller GJ, Chakrabarti RR, North WRS, et al. Haemostatic function and ischemic heart disease: principal results of the Northwick Park Heart Study. Lancet 1986; 2: 533-7.
20. Meade TW, Ruddock V, Stirling Y, Chakrabarti R, Miller GJ. Fibrinolytic activity, clotting factors, and long-term incidence of ischaemic heart disease in the Northwick Park Heart Study. Lancet 1993; 342: 1076-9.
21. Ruddock V, Meade TW. Factor VII activity and ischaemic heart disease: fatal and non-fatal events. Q J Med 1994; 87: 403-6.
22. Junker R, Heinrich J, Schulte H, van de Loo J, Assmann G. Coagulation Factor VII and the Risk of Coronry Heart Disease in Healthy Men. Arterioscler Thromb Vasc Biol 1997; 17: 1539-44.
23. Carvalho de Sousa J, Azevedo J, Soria C, Barros F, Ribeiro C, Parreira F, et al. Factor VII hyperactivity in acute myocardial thrombosis. A relation to the coagulation activation. Thromb Res 1988; 51: 165-73.
24. Orlando M, Leri O, Macioce G, Mattia G, Ferri GM. Factor VII in Subjects at Risk for Thromboembolism: Activation or Increased Synthesis? Haemostasis 1987; 17: 340-3.
25. Hoffman C, Shah A, Sodums M, Hultin MB. Factor VII activity state in coronary artery disease. J Lab Clin Med 1988; 111: 475-81.
26. Suzuki T, Yamauchi K, Matsushita T, Furumichi T, Furui H, Tsuzuki J, et al. Elevation of Factor VII Activity and Mass in Coronary Artery Disease of Varying Severity. Clin Cardiol 1991: 14; 731-6.
27. Eriksson-Berg M, Silveira A, Orth-Gomér K, Hamsten A, Schenck-Gustafsson K. Coagulation Factor VII in Middle-aged Women with and without Coronary Heart Disease. Thromb Haemost 2001; 85: 787-92.
28. Danielsen R, Önundarson PT, Thors H, Viðarsson B, Morrissey JH. Activated and Total Coagulation Factor VII, and Fibrinogen in Coronary Artery Disease. Scand Cardiovasc J 1998; 32: 87-95.
29. Folsom AR, Wu KK, Rosamond WD, Sharrett AR, Chambless LE. Prospective Study of Hemostatic Factors and Incidence of Coronary Heart Disease. The Atherosclerosis Risk in Communities (ARIC) Study. Circulation 1997; 96: 1102-8.
30. Lee AJ, Fowkes FGR, Lowe GDO, Connor JM, Rumley A. Fibrinogen, Factor VII and PAI-1 Genotypes and the Risk of Coronary and Peripheral Atherosclerosis: Edinburgh Artery Study. Thromb Haemost 1999; 81: 533-60.
31. Bladbjerg EM, Møller L, Jespersen J. Is Factor VII Protein Concentration (FVII:Ag) a Thrombotic Risk Indicator? Thromb Haemost 1998; 79: 1064-5.
32. Morrissey JH. Plasma Factor VIIa: Measurement and Potential Clinical Significance. Haemostasis 1996; 26(Suppl): 66-71.
33. Peyvandi F, Mannucci PM, Bucciarelli P, Zeinali S, Akhavan S, Sacchi E, et al. A novel polymorphism in intron 1a of the human factor VII gene (G73A): study of a healthy Italian population and of 190 young survivors of myocardial infarction. Br J Haematol 2000; 108: 247-53.
34. Girelli D, Russo C, Ferraresi P, Olivieri O, Pinotti M, Friso S, et al. Polymorphisms in the factor VII gene and the risk of myocardial infarction in patients with coronary artery disease. N Engl J Med 2000; 343: 774-80.
35. Iacoviello L, Di Castelnuovo A, de Knijff P, D´Orazio A, Amore C, Arboretti R, et al. Polymorphisms in the coagulation factor VII gene and the risk of myocardial infarction. N Engl J Med 1998; 338: 79-85.
36. Heywood DM, Ossei-Gerning N, Grant PJ. Association of Factor VII:C Levels with Environmental and Genetic Factors in Patients with Ischaemic Heart Disease and Coronary Atheroma Characterised by Angiography. Thromb Haemost 1996; 76: 161-5.
37. Doggen CJM, Manger Cats V, Bertina RM, Reitsma PH, Vandenbroucke JP, Rosendaal FR. A Genetic Propensity to High Factor VII Is not Associated with the Risk of Myocardial Infarction in Men. Thromb Haemost 1998; 80: 281-5.
38. Lievers KJA, Mennen LI, Rattink AP, Zwinderman AH, Jukema JW, Schouten EG, et al. The -323Ins10 Polymorphism for Factor VII Is Not Associated with Coronary Atherosclerosis in Symptomatic Men. Thromb Res 2000; 97: 275-80.
39. Saigo M, Abe S, Ogawa M, Biro S, Minagoe S, Maruyama I, et al. Plasma Level of Triglyceride-rich Lipoprotein Remnants Is Closely Associated with the Activation of Coagulation Factor VII in Patients with Myocardial Infarction. Thromb Res 2000; 100: 9-17.
40. Larsen LF, Bladbjerg E-M, Jespersen J, Marckmann P. Effects of Dietary Fat Quality and Quantity on Postprandial Activation of Blood Coagulation Factor VII. Arterioscler Thromb Vasc Biol 1997; 17: 2904-9.
41. Aledort LM. Recombinant Factor VIIa Is a Pan-hemostatic Agent? Thromb Haemost 2000; 83: 637-8.