

Hrörnunarsjúkdómar í heila - oxavarnarensím og kopar Kynning á rannsóknum
Ágrip
Inngangur: Hrörnunarsjúkdómar í miðtaugakerfi eiga langflestir það sameiginlegt að í þeim á sér stað samsöfnun og útfelling á próteinum í taugafrumum eða utan við þær hver sem orsökin kann að vera. Talið er að efnabreytingar, sem eru undanfari samsöfnunar og útfellinga, séu skaðvaldurinn fremur en útfellingarnar sjálfar. Efnabreytingar þessar leiða að öllum líkindum til myndunar á skaðlegum súrefnisfríhópum. Oxavarnir líkamans, sem bæði taka til sértækra efna og oxavarnarensíma, vinna gegn þessu ferli og því hefur þeirri tilgátu verið varpað fram að veiklaðar oxavarnir séu sameiginlegur þáttur í meingerð hrörnunarsjúkdóma í miðtaugakerfi, hvort sem er í mönnum eða öðrum spendýrum.
Aðferðir: Magn kopars og virkni tveggja oxavarnandi ensíma sem innihalda kopar, cerúlóplasmíns og súperoxíðdísmútasa 1 (SOD1), var ákvarðað í blóðinu. Gerðar voru tvenndarrannsóknir er tóku til Alzheimer sjúkdóms, Parkinson sjúkdóms, hreyfitaugungahrörnunar og sjúklinga með Downs heilkenni auk sjúklinga með einhverfu. Einnig var gerð rannsókn á sauðfé á mismunandi svæðum með mismunandi líkum á riðusmiti. Í þeirri rannsókn var að auki ákvörðuð virkni glútatíonperoxídasa sem er selenríkt oxavarnarensím og magn mangans ákvarðað í blóðinu.
Niðurstöður: Oxunarvirkni cerúlóplasmíns og virkni SOD1 var marktækt minni í Alzheimer sjúkdómi án þess að rekja mætti það til vöntunar á kopar. Í Parkinson sjúkdómi var virkni cerúlóplasmíns einnig marktækt minni og virkni bæði cerúlóplasmíns og SOD1 minnkaði marktækt með sjúkdómslengd enda þótt kopar væri innan eðlilegra marka. Í hreyfitaugungahrörnun var breytileiki einstakra mælingargilda cerúlóplasmíns og SOD1 marktækt öðruvísi en ekki var munur á meðaltölugildum. Í einstaklingum með Downs heilkenni sem voru 40 ára og eldri og því komnir á þann aldur að Alzheimerlíkra breytinga er að vænta í heilanum, var virkni SOD1 og sértæk virkni cerúlóplasmíns (virkni í hlutfalli við magn) marktækt minni en í yngri hluta hópsins. Í einhverfu, sem einkennist af þroskahefti fremur en vaxandi hrörnunareinkennum, var hins vegar enginn munur á sjúklingum í samanburði við heilbrigða einstaklinga. Niðurstöður rannsókna á sauðfé bentu til þess að samhengi gæti verið milli aukinnar hættu á riðusmiti og minnkandi virkni glútatíonperoxídasa og hugsanlega einnig minnkandi SOD1 virkni. Engin marktæk tengsl voru að því er virtist milli aukinnar hættu á riðusmiti og breytinga á virkni cerúlóplasmíns. Aukin hætta á riðusmiti varð ekki tengd við litla þéttni kopars eða mikla þéttni mangans í blóði fjárins.
Umræða: Rannsóknirnar benda til þess að oxavarnir séu veiklaðar í þeim fjórum hrörnunarsjúkdómum í miðtaugakerfi manna sem rannsakaðir voru þótt klínísk mynd þeirra sé ærið ólík, en greina mátti minnkaða eða afbrigðilega virkni oxavarnandi koparensíma í blóði við alla þessa sjúkdóma. Í ástandi sem telst vera þroskahefti (einhverfa) og er án virkra hrörnunarbreytinga eða útfellinga í heila er ekki að finna slíkar breytingar. Niðurstöðurnar styðja því þá tilgátu að veiklaðar oxavarnir séu sameiginlegur þáttur í meingerð þessara sjúkdóma. Varðandi sauðfé benda niðurstöður einnig til þess að veiklun sé í oxavörnum samfara auknum líkum á riðusmiti þótt það verði að líkindum einkum tengt minnkaðri virkni glútatíonperoxídasa.
Hrörnunarsjúkdómar í heila
Algengustu hrörnunarsjúkdómar í heila manna, Alzheimer sjúkdómur og Parkinson sjúkdómur, eru aldursháðir, þótt þeir séu ekki aldursbundnir (1). Þetta merkir að í báðum tilvikum eykst algengi með hækkandi aldri (sér í lagi eftir 60 ára aldur), en báðir sjúkdómar eru jafnframt vel þekktir og greindir í yngra fólki. Svo var einmitt um fyrsta tilfellið af Alzheimer sjúkdómi sem komst á bækur og var lýst af þýska lækninum Alois Alzheimer 1907 (2).
Þegar sjúkdómar þessir byrja á tiltölulega ungum aldri, og í vissum tilvikum einnig síðar, má oft greina arfbundnar genabreytingar sem eru ákvarðandi fyrir sjúkdómsmyndina (meingerðina). Sem dæmi um þetta má nefna að þekktir eru allmargir gallar í þremur genum er leiða til snemmkomins Alzheimer sjúkdóms (3). Slíkar genabreytingar eru alls ráðandi að talið er við uppkomu sjaldgæfra pólýglútamínhrörnunarsjúkdóma í miðtaugakerfinu. Þekktastur þeirra er væntanlega Huntington sjúkdómur (4, 5). Í öðrum tilvikum geta arfbundnar genabreytingar, eða breytileiki í gerð gena, verið sjúkdómshvetjandi. Þannig háttar sambandi Apo-E og síðkomins Alzheimer sjúkdóms (6) og líkur benda einnig til áhrifa mismunandi arfgerðar á uppkomu Parkinson sjúkdóms (7). Í langflestum tilvikum eru orsakir þessara sjúkdóma þó lítt kunnar, en kunna að tengjast utanaðkomandi efnum eða ákomum eða samspili þeirra og erfða. Hvort sem Alzheimer sjúkdómur eða Parkinson sjúkdómur byrjar fyrr eða síðar, eða er arfbundinn eða ekki, eru sjúkdómsmyndin og sjúkdómseinkennin í stórum dráttum hin sömu. Athygli vekur og að þessir sjúkdómar geta hvarfast saman, það er að segja sjúklingar geta fyrst fengið einkenni um annan sjúkdóminn, en síðar einkenni um hinn.
Við Alzheimer sjúkdóm ber í upphafi mest á minnisglöpum, einkum skerðingu á nýminni og verknaðarminni, og verkstoli með minnkandi getu til skipulagningar og framkvæmda á flóknari athöfnum daglegs lífs. Þessi einkenni ásamt ýmsum öðrum eru ríkjandi við það sjúkdómsástand sem nú er nefnt heilabilun (dementia). Heilabilunarsjúkdómar eru ýmsir þekktir, en Alzheimer sjúkdómur er þeirra langalgengastur (1, 8).
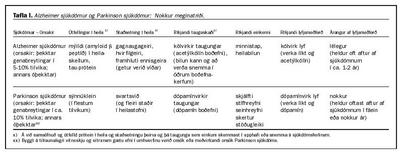
Við Parkinson sjúkdóm eru ríkjandi truflanir í viljabundnum hreyfingum sem er langoftast að rekja til hrörnunar í dópamínvirkum taugungum í svartsviði (substantia nigra) í miðheila (9). Í töflu I er að finna nokkur meginatriði um Alzheimer og Parkinson sjúkdóma.
Í dýrum (sauðfé, nautgripum og fleiri) þekkjast aðrir hrörnunarsjúkdómar í miðtaugakerfi er nefnast einu nafni príonsjúkdómar. Þeir þekkjast raunar í nokkrum mæli í mönnum einnig (Creutzfeldt-Jacobs sjúkdómur og fleiri). Líkt og við Alzheimer sjúkdóm og Parkinson sjúkdóm kunna breytingar á ákvarðandi genum eða breytileiki í gerð gena að ráða uppkomu príonsjúkdóma eða hvetja til uppkomu þeirra í vissum tilvikum. Orsakir príonsjúkdóma eru þó, líkt og á við hina tvo hrörnunarsjúkdómana, oftast lítt kunnar eða óljósar (1).
Af príonsjúkdómum í dýrum er sauðfjárriða, oft stytt í riða (scrapie), vel þekkt hér á landi. Eru þá skemmdir í miðtaugakerfinu einkum í heilastofni og litla heila (10). Kúariða, riða í nautgripum ("mad cow disease"), er ekki þekkt hér á landi, en hefur valdið miklum usla og skelfingu í Bretlandi og á meginlandi Evrópu. Skelfing vegna kúariðu hefur mótast af því að nýtt afbrigði af Creutzfeldt-Jakobs sjúkdómi, sem einkum leggst á ungt fólk, er af ýmsum talinn hafa borist í menn með neyslu kjöts af sýktum nautgripum (1, 11).
Öfugt við Alzheimer sjúkdóm og Parkinson sjúkdóm teljast príonsjúkdómar jafnframt til smitsjúkdóma. Þetta á jafnt við sauðfjárriðu, kúariðu og Creutzfeldt-Jakobs sjúkdóm í mönnum. Smithættan er fyrst og fremst meðal einstaklinga innan sömu tegundar. Reynsla er samt ótvírætt fyrir því að möguleikar eru á smitun milli einstaklinga mismunandi dýrategunda. Engar vísbendingar eru um að sauðfjárriða hafi nokkru sinni borist í menn, né tengist uppkomu Creutzfeldt-Jakobs sjúkdóms eða annarra príonsjúkdóma í mönnum (1, 11). Sjá einnig viðbæti.
Við sauðfjárriðu, kúariðu, Creutzfeldt-Jakobs sjúkdóm og aðra príonsjúkdóma má oftast greina í smásjá eins konar göt í heilafrumum líkt og svampur væri. Þessar breytingar kallast á erlendum málum "spongiform vacuolation" (1, 12). Eru príonsjúkdómar því stundum nefndir einu heiti "transmissible spongiform encephalopathies" (TSE), þar eð um smitsjúkdóma er að ræða.
Enda þótt verulegur munur sé milli fyrrnefndra hrörnunarsjúkdóma í miðtaugakerfi bæði að því er varðar sjúkdómseinkenni, gerð vefjaskemmda (sjúkdómsmyndar) og staðsetningar í miðtaugakerfinu, er samt vaxandi skilningur á því að þessir hrörnunarsjúkdómar, svo og Huntington sjúkdómur og ýmsir aðrir ónefndir hrörnunarsjúkdómar, séu allir af sams konar rót (1, 12). Hér breytir að því er virðist engu þótt príonsjúkdómar teljist smitsjúkdómar, en aðrir hrörnunarsjúkdómar í miðtaugakerfi ekki. Í öllum tilvikum á sér stað samsöfnun (aggregation) og útfelling á próteinum inni í taugafrumum eða utan við þær, hvort sem þessar sjúklegu breytingar má rekja til þekktra breytinga á ákvarðandi genum eða ekki. Í öllum þekktum tilvikum virðist ennfremur vera um að ræða starfræn prótein sem ætla má að gegni nauðsynlegu hlutverki í starfi miðtaugakerfisins, en breytast af ýmsum sökum í óstarfhæfan próteinmassa samfara vefjaskemmandi verkun. Beinn skortur á starfrænum próteinum kann vissulega einnig að móta sjúkdómsmyndina, að minnsta kosti við suma arfbundna hrörnunarsjúkdóma (5).
Breytingin úr starfrænum próteinum í síður starfræn eða beinlínis óstarfhæf prótein er fólgin í formbreytingu (afformun, umformun), sem ekki er samfara meiriháttar efnafræðilegri breytingu. Þetta merkir að lega próteinanna breytist í rúminu og leysanleiki þeirra minnkar án þess að samsetning þeirra breytist eða þurfi að breytast marktækt. Þetta stuðlar að samsöfnun óeðlilega margra próteinsameinda og útfellingu í óstarfhæfan massa. Við eðlilegt ástand eru ýmis ráð tiltæk til þess að tryggja að starfræn prótein "rúllist rétt" og fái við það rétt starfrænt form eða að þeim sé að öðrum kosti sundrað og eytt. Þessi kerfi geta greinilega bilað við þá hrörnunarsjúkdóma sem hér um ræðir (13). Taugafrumur virðast ennfremur vera sérstaklega viðkvæmar gegn þeim eiturhrifum, sem af þessum breytingum hljótast (12).
Í Alzheimer sjúkdómi (tafla 1) og mörgum fleiri fátíðari hrörnunarsjúkdómum í miðtaugakerfi eru oftast áberandi útfellingar á svokölluðu tau-próteini, en það er í eðlilegu ástandi nauðsynlegur hluti af burðarvirki frumnanna (cytoskeleton). Annað afbrigðilegt prótein í Alzheimer sjúkdómi og nokkrum fleiri hrörnunarsjúkdómum er mýildi (amyloid b peptíð; Ab). Það er aðaluppistaðan í heilaskellum (cerebral plaques; senile plaques) sem oftast eru áberandi í heila Alzheimer sjúklinga og í fólki með Downs heilkenni eftir miðjan aldur (sjá síðar). Mýildi má skoða sem samsöfnun og útfellingu á sérstakri klofningsafurð úr mun stærra forstigspróteini (amyloid precursor protein; APP) sem er í frumuhimnu og nauðsynlegt er talið fyrir starfsemi taugafrumna og sennilega einnig annarra frumna. Aðalklofningsafurð (Ab1-40) þessa forstigspróteins myndar ekki mýildi. Við Alzheimer sjúkdóm og í Downs heilkenni myndast óhæfilega mikið af áðurnefndri mýildisgefandi klofningsafurð (Ab1-42) úr forstigspróteininu, hugsanlega vegna of mikillar virkni tiltekins próteasa, b-sekretasa (14). Allt bendir til þess að við báða þessa sjúkdóma sé myndun þessarar mýildisgefandi klofningsafurðar og uppkoma heilaskellna ennfremur með einum eða öðrum hætti undanfari útfellingar á tau-prótíni í heilanum (15).
Í Parkinson sjúkdómi (tafla 1) eru svokallaðar Lewys-agnir langoftast áberandi í frumum í svartsviði (1). Í þessum ögnum hafa samsafnast í mýildislíka myndun um tveir tugir efna, en mest er af efni sem nefnist sýnnúklein (a-synnuclein). Sýnnúklein er talið hafa, líkt og klofningsafurðir úr áðurnefndu forstigspróteini, hlutverki að gegna í taugastarfseminni. Þegar Lewys-agnir myndast í frumum í heilaberki koma fram einkenni heilabilunar, svokallaður Lewy sjúkdómur (Dementia with Lewy bodies) (16).
Enn er að nefna hið eðlilega príonprótein (PrPc) sem umformast (afformast) í príonsjúkdómum í sjúklegt príonprótein (PrPsc). Það safnast í mismunandi miklum mæli í skellur í holum taugavefnum sem geta minnt á heilaskellur við Alzheimer sjúkdóm (1). Eðlilegt príonprótein inniheldur kopar og virðist geta bundið hann og þannig meðal annars forðað skemmdum af völdum kopars í miðtaugakerfinu. Það hefur einnig oxavarnandi (antíoxídatíf) verkun (17). Athyglisvert er að sýnnúklein inniheldur einnig kopar. Kopar virðist og marktækt geta hvatað útfellingu á mýildi, sem myndast út frá forstigspróteininu eins og áður ræðir (sjá á eftir).
Rannsóknir síðustu ára benda samt til þess að samsöfnuð eða útfelld prótein í óstarfrænan massa í taugavefnum séu í sjálfu sér tiltölulega lítt skaðleg. Skaðsemin felst að öllum líkindum einkum í óverulegum efnabreytingum á vissum, leysanlegum starfrænum peptíðum eða próteinum (eða starfrænum próteinum réttbundnum við frumuhimnur), hvort sem þau eru klofningsafurðir úr stærri forstigspróteinum eða ekki, sem svo eru undanfari samsöfnunar og útfellingar (18, 19). Við efnabreytingar þessar er talið að súrefnisfríhópar myndist í próteinunum sem oxi fitur (lípíða) í frumuhimnum og geri þær með því lekar. Aðrir staðir í frumunum sem gætu skemmst og haft afdrifaríkar afleiðingar eru ensím í orkukornum (mitochondria) eða víðar og "próteinkvarnir" (próteasóm) sem eyða próteinum, ekki síst afformuðum próteinum í frumum (1, 12, 13). Að þessu loknu, eða ef til vill samtímis, verði síðan samsöfnun á próteinsameindum og útfelling í óstarfhæfan massa. Nýlegar rannsóknir benda sömuleiðis til þess að frumuhimnur sem skemmst hafa vegna oxunar geti hvatað myndun mýildis mun meira en heilar frumuhimnur (20).
Af þessum fyrirbærum má meðal annars draga tvær ályktanir. Í fyrsta lagi endurspeglar massi útfelldra próteina ekki beinlínis skemmdirnar í taugavefnum heldur eru þær fremur vitni um "kulnað endastig". Í öðru lagi er ljóst að bilaðar oxavarnir líkamans gætu skipt meginmáli við uppkomu þessara sjúkdóma, hvort sem þeir byrja á ungum aldri eða gömlum. Síðastnefnda atriðið var kveikjan að rannsóknum okkar sem hér greinir frá. Fyrst verður þó vikið lítillega að oxun, oxavörnum og oxavarnarensímum.
Oxun og oxavarnarensím
Orkunám er nauðsyn öllum lífverum. Í öllum æðri lífverum fer orkunámið fram í svokölluðum orkukornum (mitochondria) í frumunum. Orkunám byggist á oxun sem felur í sér hvörf (afoxun) súrefnis úr andrúmsloftinu við orkugefandi fæðuefni. Súrefni afoxast við það stig af stigi í vatn. Samtímis losnar orka sem bundin er í orkuríkum fosfatsamböndum (adenósínþrífosfat, kreatínfosfat) og eru eins og allsherjar hreyfanlegar orkustöðvar fyrir alla líkamsstarfsemina. Þegar súrefni afoxast tekur það í sig, stig af stigi, neikvæða hleðslu (tekur í sig rafeindir) og binst endanlega vetni og myndar vatn. Við þessi efnahvörf myndast ýmsar milliafurðir. Geta þær hæglega skaðað frumur ef myndun þeirra er úr hófi eða ummyndun þeirra í vatn er ekki sem skyldi og ekki gildir það síst um taugavef (21, 22).
Við ummyndun í vatn (H2O) tekur ein sameind súrefnis (O2) í sig alls fjórar rafeindir, hverja á fætur annarri. Fyrsta afurðin er .O2_, oft nefnd súperoxíðanjón-fríhópur (tafla II, liður 1).
Með hugtakinu fríhópur (free radical) er átt við að hlutaðeigandi efni hafi fengið (eða gefið frá sér) staka rafeind. Við það verður efnishópurinn hvarfgjarn, "frjáls" eða "frír" til efnahvarfa í mismunandi miklum mæli. Samtímis leitast hópurinn við að taka til sín (eða gefa frá sér) aðra rafeind og umbreytast í stöðugra efnasamband. Ef súrefni á í hlut, kallast fríhópar þess einu nafni súrefnisfríhópar (21).
Næsta skref í ummyndun súrefnis í vatn er myndun súrvatns (vetnissúperoxíðs; vetnisperoxíðs), H2O2, út frá súperoxíðanjónfríhópi, sem fyrr greinir. Súrvatn (nafnið er dregið af því að í sameindinni er einu súrefnisatómi fleira en í sameind vatns) er að vísu ekki fríhópur. Það hvarfast hins vegar greiðlega áfram í .OH, hýdroxífríhóp, og hýdroxíanjón (OH_), sem gengur í samband við vetni og myndar eina sameind af vatni. Hýdroxífríhópurinn hvarfast svo áfram í hýdroxíanjón, sem binst vetni og myndar aðra sameind vatns (tafla II, liðir 2-4).
Af framansögðu er ljóst að vegna afoxunar á einni sameind af súrefni myndast við eðlilegar aðstæður tvær sameindir af vatni (tafla II). Ef truflun verður á þessu efnaferli er aukin hætta á myndun súrefnisfríhópa með eftirfarandi vefjaskemmdum sem tengst geta uppkomu margra sjúkdóma, þar á meðal uppkomu hrörnunarsjúkdóma í miðtaugakerfinu. Til dæmis má nefna að of mikil myndun á .O2_ (súperoxíðanjónfríhóp) eða of hæg ummyndun í H2O2 (súrvatn) getur valdið því að þessi tvö efni hvarfist saman. Myndast þá .OH, hýdroxífríhópur sem hæglega getur skemmt frumuhimnur, kjarnaprótein og fleira þar í nánd sem hann myndast (21). Hvörf .O2_ við NO (níturoxíð) sem myndast víða í líkamanum geta einnig leitt til myndunar á .OH (23). Í þessu sambandi vekur athygli að mýildi, og ekki síður forstigspróteinið, geta greiðlega bundið málma á borð við járn og kopar sem hvata myndun hýdroxífríhópa, svo og zink og fleiri málma.
Raunar virðist mýildi ekki samsafnast og falla út að marki nema eftir bindingu við málma, einkum kopar og zink (24). Mýildi (og einnig forstigspróteinið) hafa ekki aðeins mikla sækni í kopar og járn heldur geta og afoxað tvígildan kopar eða þrígilt járn í eingildan kopar eða tvígilt járn. Samfara þessum hvörfum myndast súrvatn (H2O2) út frá súrefni (O2). Eingildur kopar eða tvígilt járn geta því næst marktækt hvatað myndun á .OH (hýdroxífríhóp) út frá H2O2 sem hefur staðbundna vefjaskemmandi verkun eins og áður segir (tafla II, liður 3). Eingildur kopar eða tvígilt járn sem myndast samkvæmt framansögðu, kunna því að vera mikils ráðandi bæði um vefjaskemmdir af völdum mýildis svo og um samsöfnun þess og útfellingu í taugavef (24).
Til þess að tryggja greiða afoxun súrefnis í vatn og bægja frá hættu, ef eitthvað stefnir í að fara úrskeiðis í því ferli, ræður líkaminn yfir ýmsum ensímkerfum, sem einu nafni nefnast oxavarnarensím. Ennfremur nýtir hann andoxunarefni, svo sem tókóferól, úbíkínón og fleiri. Þetta tvennt eru uppistaðan í oxavörnum (vörnum gegn oxunarskemmdum) í líkamanum (21, 22).
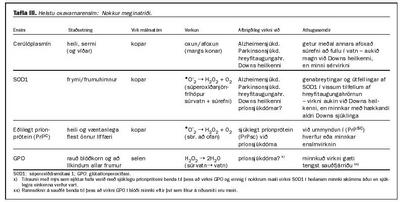
Meðal helstu oxavarnarensíma eru súperoxídasar, glútatíonperoxídasi og cerúlóplasmín. Súperoxídasar eru járn-, kopar- eða manganensím, cerúlóplasmín er koparensím og glútatíonperoxídasi er selenensím. Þetta merkir að ýmist járn, kopar, mangan eða selen eru í svokölluðum virkum sætum í ensímsameindunum og er það ásamt öðru nauðsynlegt fyrir virkni þeirra.
Helstu súperoxídasar í líkamanum eru tveir, skammstafað SOD1 og SOD2. SOD1 er koparensím (inniheldur einnig zink) og er að finna í frymi langflestra frumna eða í einhverjum mæli í frumuhimnum. SOD2 er lífsnauðsynlegt manganensím sem er í orkukornum frumna og ekki ræðir hér frekar. Auk þess hefur hið eðlilega príonprótein SOD virkni, en það er bundið við frumuhimnur bæði í miðtaugakerfinu og utan og er koparensím. Verkun þessara ensíma er að hvata breytinguna á .O2_ í H2O2 og súrefni (tafla III).
Glútatíonperoxídasi er í rauðum blóðkornum og að líkindum í nánast öllum frumum. Selen er ákvarðandi efni í sameindinni eins og áður segir, en afoxað glútatíon er hjálparefni við ensímhvörfin. Glútatíonperoxídasi, oft skammstafað GPO, breytir súrvatni, H2O2, í vatn (H2O). GPO verkar þannig "aftan" við SOD (tafla III). Að auki eyðir GPO einnig öðrum peroxíðum, til dæmis í fitum í frumuhimnum (fitur í frumuhimnum sem tekið hafa í sig aukalegt súrefni vegna utanaðkomandi oxunar), og skiptir þannig mjög verulegu máli í þá veru að varna frumuskemmdum.
Cerúlóplasmín finnst í sermi og er einnig bundið frumuhimnum í miðtaugakerfinu. Það inniheldur mörg koparatóm og starfar að nokkru við að flytja kopar um líkamann. Cerúlóplasmín er einnig mjög öflugt ensím, hvort sem er eftir atvikum til oxunar eða afoxunar, og það getur eitt sér afoxað súrefni fullkomlega í vatn (tafla III). Þá skiptir oxunarvirkni cerúlóplasmíns meginmáli í þá veru að tryggja rétt oxunarstig á járni (rétt hlutfall milli tvígilds og þrígilds járns) og með því óbeint að tryggja flutning þess um líkamann samanber á undan. Það eitt dregur einnig úr líkum á því að oxunarskemmdir verði í líkamanum (25, 26). Cerúlóplasmín er ennfremur eitt svokallaðra álagspróteina í líkamanum og eykst að magni við ýmis konar álag, ekki síst samfara aukinni súrefnisneyslu, svo sem við bólgur af hvers kyns toga (27).
Hugmyndafræðin bak við rannsóknir okkar var að kanna hvort breytingar á magni eða virkni cerúlóplasmíns eða virkni SOD1 yrði vart í blóði sjúklinga með Alzheimer sjúkdóm eða Parkinson sjúkdóm í þeim mæli að það skipti máli við greiningu þessara sjúkdóma eða hefði gildi við mat á framvindu þeirra. Með því að bæði þessi ensím eru koparensím var magn kopars einnig ákvarðað í blóðinu. Við víkkuðum svo rannsóknarsviðið með því að taka sýni úr sjúklingum með hreyfitaugungahrörnun (amyotrophic lateral sclerosis) og Downs heilkenni. Í þeirri rannsókn vakti fyrir okkur að kanna hvort sömu eða svipaðar ensímbreytingar væru í blóði eldri sjúklinga með Downs heilkenni, sem sýna oftast merki um heilabilun, og eru í blóði Alzheimer sjúklinga. Þá tókum við til samanburðar sýni úr blóði fólks með einhverfu (autism). Einhverfa er, öfugt við Alzheimer sjúkdóm, Parkinson sjúkdóm, hreyfitaugungahrörnun og Downs heilkenni með einkennum um heilabilun, talin vera sjúkdómsástand sem einkennist af þroskahefti eða misþroska í miðtaugakerfinu fremur en hrörnun og dauða á áður fullþroskuðum frumum. Loks var rannsóknarsviðið til samanburðar víkkað út til þess að ná einnig til sauðfjár og líkum á riðusmiti í því. Við riðurannsóknirnar var af sökum sem síðar ræðir lögð sérstök áhersla á að ákvarða virkni glútatíonperoxídasa og magn mangans auk kopars í blóðinu. Verður í lokin vikið að þeim rannsóknum.
Kopar og oxavarnarensím í Alzheimersjúkdómi og Downs heilkenni
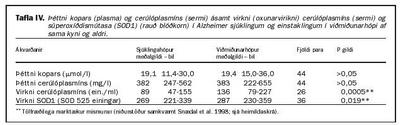
Gerðar voru tvenndarrannsóknir (case control studies) á allt að 40 tvenndum eða pörum þar sem annars vegar voru sjúklingar með skilgreindan Alzheimer sjúkdóm, en hins vegar einstaklingar af sama kyni og aldri, sem töldust heilbrigðir samkvæmt nánari skilmerkjum. Lagt var fyrir sjúklingana einfalt vitrænt próf (Mini Mental State Examination = MMSE). Töldust þeir að jafnaði hafa meðalþungan sjúkdóm (moderate) með að meðaltali 16 stig af 30 mögulegum. Helstu niðurstöður eru sýndar í töflu IV.
Niðurstöður þessara rannsókna voru á þá leið að magn kopars var innan þeirra marka sem telst eðlilegt. Oxunarvirkni cerúlóplasmíns og SOD1 virknin var hins vegar marktækt minni í sjúklingunum en í samanburðarhópnum. Enginn munur var á magni cerúlóplasmíns í þessum tveimur hópum. Merkir það því að sérvirkni cerúlóplasmíns (virkni í hlutfalli við massa) var miklum mun minni í sjúklingunum en í samanburðarhópnum (ekki sýnt í töflu IV). Ekki fannst að marktæk tengsl væru milli breytinga á virkni cerúlóplasmíns og SOD1 virkni, né var í þessari rannsókn unnt að tengja breytingar á ensímvirkni við aldur eða lengd sjúkdómsins eða niðurstöður í MMSE prófi (6).
Margir af þeim sjúklingum sem tóku þátt í rannsókninni eru nú, að fimm til sex árum liðnum, enn á lífi. Er til athugunar að fá þá til rannsóknar á ný til þess að kanna hvort breytingar á virkni oxavarnarensíma í blóði hafi eitthvert forsagnargildi um sjúkdómsferilinn. Þetta er sérlega áhugavert í ljósi þess að í velgerðri tvenndarrannsókn var aukin fituoxun (lipid peroxidation) í gagnaugageira Alzheimer sjúklinga post mortem í samanburði við fólk í viðmiðunarhópi. Samtímis var virkni SOD1 og katalasa (klýfur H2O2) einnig marktækt minni, en ekki virkni glútatíonperoxídasa (28).
Downs heilkenni er meðfæddur ágalli eða sjúkdómur sem langoftast er að rekja til þrísætu (trisomia) á litningi 21, en sjaldnast er arfgengur. Slíkt ofmagn gena ber með sér margháttað þroskahefti og samfara því eru sjúklegar breytingar í mörgum líffærum. Í þessum einstaklingum er meðal annars eitt aukalegt gen fyrir forstigsprótein (APP) mýildis, en mýildi er uppistaðan í heilaskellum eins og áður er vikið að. Svo sem vænta má eru slíkar heilaskellur langoftast greinanlegar hjá sjúklingum með Downs heilkenni þegar fyrir fertugt og heilabilunareinkenni eru algeng upp úr fimmtugu (29).
Þessir sjúklingar hafa einnig þrjú gen fyrir SOD1, þar eð genið sem skráir gerð þessa ensíms er sömuleiðis á litningi 21. Er því SOD1 virknin hjá þeim að jafnaði upp undir 50% meiri en venjulegt er. Umdeilt er hvort þessi aukning á SOD1 virkni skiptir yfirleitt máli fyrir ríkjandi einkenni í Downs heilkenni eða ekki. Frá okkar sjónarmiði var hins vegar áhugavert að vita hvort virknin minnkaði með uppkomu Alzheimerlíkra breytinga í heila á rosknum aldri.
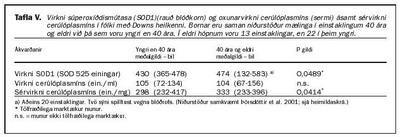
Gerð var tvenndarrannsókn á 35 tvenndum (pörum) líkt og áður er lýst og í öllum tilfellum nema einu var klínísk greining á Downs heilkenni staðfest með litningagreiningu. Að þessu loknu voru niðurstöður brotnar upp eftir aldri. Þær sýndu að í eldri hluta hópsins (40 ára eða eldri) var SOD1 virknin marktækt minni en í yngri hópnum (tafla V). Sérvirkni cerúlóplasmíns (virkni í hlutfalli við massa) var einnig marktækt minni hjá þeim sem voru 40 ára eða eldri en hjá þeim yngri. Magn cerúlóplasmíns (og þar með magn kopars) fór hins vegar marktækt hækkandi með hækkandi aldri (ekki sýnt í töflu V). Oxunarvirkni cerúlóplasmíns hélst því af þessum sökum í heild óbreytt og var hin sama bæði hjá þeim sem voru 40 ára eða eldri og hjá þeim sem voru yngri (tafla V). Það var til baga í þessari rannsókn hve tiltölulega fáir í rannsóknarhópnum voru á sextugsaldri. Okkar skoðun er sú að þessar breytingar hefðu orðið enn marktækari ef fleiri hefðu verið í elsta aldurshópnum (26).
Að öllu samanlögðu teljum við því að við Alzheimer sjúkdóm megi greina truflun á starfi oxavarnandi koparensíma í blóði sem ekki er að rekja til skorts á kopar. Hliðstæðar breytingar verða á starfsemi þessara ensíma hjá sjúklingum með Downs heilkenni á rosknum aldri, eða um svipað leyti og Alzheimerlíkra breytinga í heila verður vart hjá þeim. Rannsaka þarf nánar hvort eða hvernig sjúkdómsferillinn mótast af minnkaðri virkni þessara ensíma. Í þessu efni vekur athygli að virkni cerúlóplasmíns og SOD1 breyttist óháð hvor annarri hjá Alzheimer sjúklingum. Aukin myndun á cerúlóplasmíni í rosknum Downs sjúklingum er trúlega álagseinkenni. Í Downs-rannsókninni vekur einnig athygli að sérvirkni cerúlóplasmíns minnkaði marktækt á rosknum aldri, þrátt fyrir marktækt aukna myndun á cerúlóplasmíni með hækkandi aldri.
Kopar og oxavarnarensím í Parkinson sjúkdómi og hreyfitaugungahrörnun
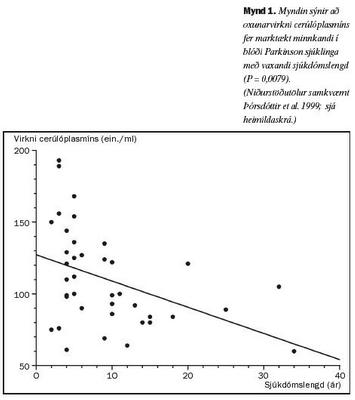
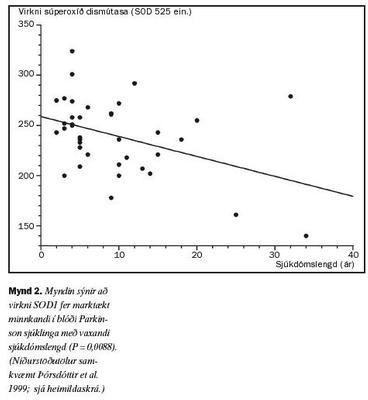
Rannsókn var gerð á 40 tvenndum einstaklinga. Einstaklingar í öðrum helmingi voru með skilgreindan Parkinson sjúkdóm, en í hinum helmingi voru heilbrigðir einstaklingar samkvæmt nánari skilmerkjum. Kopar var innan eðlilegra marka í blóði þessara sjúklinga. Í samanburði við viðmiðunarhópinn var hins vegar bæði magn, oxunarvirkni og sértæk virkni cerúlóplasmíns marktækt minni í sermi sjúklinganna. SOD1 virknin var ekki marktækt minni í rauðum blóðkornum hjá sjúklingunum, en minnkaði marktækt í hlutfalli við sjúkdómslengd (mynd 1). Oxunarvirkni cerúlóplasmíns fór einnig marktækt minnkandi í hlutfalli við sjúkdómslengdina (mynd 2). Í báðum tilvikum minnkaði ensímvirknin óháð aldri og virkni cerúlóplasmíns og SOD1 breyttist óháð hvor annarri.
Í þessari rannsókn var ennfremur rannsakað hvort marktækar truflanir væru í járnbúskap Parkinson sjúklinga. Var þannig ákvarðað járn í sermi, serum ferritín, transferrín, mettun transferríns og járnbindigeta. Var þetta gert vegna þess að fyrri athuganir höfðu bent til þess að truflanir væru á járnbúskap hjá sjúklingum með Parkinson sjúkdóm. Ekkert slíkt fannst í okkar rannsókn (25).
Sérstaka athygli vakti í þessari rannsókn að virkni beggja ensíma minnkaði marktækt, óháð hvor annarri, með lengd sjúkdómsins. Engir sjúklinganna höfðu önnur marktæk einkenni né liðu af lélegri næringu sem skýrt gæti þessa niðurstöðu og eru margir þeirra enn á lífi. Hafin er nú ný rannsókn til þess að kanna hvort minnkandi cerúlóplasmínvirkni eða SOD1 virkni, önnur eða hvortveggja, tengist sjúkdómsferlinum svo að marktækt sé. Ef svo er gæti ákvörðun á virkni annars eða beggja ensíma haft forspárgildi um sjúkdómsferilinn. Járnútfellingar eru þekktar í heila hjá Parkinson sjúklingum. Orsakir þessa kunna að vera margar, meðal annars truflun í virkni cerúlóplasmíns (sjá á undan).
Hugsanlegt er ennfremur að truflun á virkni cerúlóplasmíns geti tengst uppkomu Parkinson sjúkdóms í ríkari mæli en nú er þekkt. Við höfum þess vegna hafið rannsókn á því hvort tengsl kunni að vera milli Wilson sjúkdóms, þar sem eru þekktar truflanir í koparbúskap, og Parkinson sjúkdóms.
Efni í umhverfinu geta sannanlega valdið skemmdum á dópamínvirkum taugungum í svartsviði. Ef áverkunin er nægilega mikil veldur það sjúkdómsástandi í mönnum og dýrum er líkist Parkinson sjúkdómi (Lewy skellur vantar þó að jafnaði). Best rannsakað slíkra efna er MPTP (1-metýl-4-fenýl-1,2,3,6 tetrahýdrópýridín). MPTP oxast í glia frumum (bandvefsfrumum í miðtaugakerfinu), einkum í stirnufrumum (astrocytar), fyrir tilstilli mónóamínóxidasa B í MPP+ (1-metýl-4-fenýlpýridín), sem því næst er ferjað inn í dópamínvirka taugunga með sömu ferju og flytur dópamín inn í taugafrumur á ný við endurupptöku. Genabreyttar mýs sem hafa marktækt meiri SOD1 virkni en venjulegar mýs eru mun síður næmar gegn skemmandi verkun af völdum MPP+ en þær. Þetta rennir eindregið stoðum undir þá skoðun að oxunarskemmdir séu valdar að sjúkdómseinkennunum og eykur á vægi endurrannsóknar á Parkinson sjúklingum er að framan ræðir (30-32).
Hreyfitaugungahrörnun er illvígur og oftast mjög banvænn sjúkdómur sem stafar af hrörnun í hreyfitaugungum í heilastofni og mænu og er oftast samfara próteinútfellingum í frumunum. Í 5-10% tilfellna er sjúkdómurinn arfbundinn og í hluta af þeim tilfellum er um að ræða breytingu á SOD1 geninu svo og í vissum tilvikum þegar sjúkdómurinn er ekki arfgengur. Í þessum tilfellum eru útfellingar í frumunum oft aðallega gerðar úr SOD1 próteini en í flestum öðrum tilfellum eru útfellingar í taugafrumum af öðrum uppruna (33).
Niðurstöðutölur úr rannsókn á sjúklingum með hreyfitaugungahrörnun, sem var tvenndarrannsókn með sama hætti og áður, bentu til þess að breytileiki (equality of variance) sértækrar cerúlóplasmínvirkni og SOD1 virkni væri marktækt öðruvísi í sjúklingunum en var í samanburðarhópnum, enda þótt enginn munur væri á miðtölugildum. Þéttni kopars í plasma var hin sama í báðum hópum (34). Þessar niðurstöður gefa því vísbendingu um að einnig í þessum hrörnunarsjúkdómi sé truflun á starfi oxavarnarensíma í blóði sem innihalda kopar þótt koparmagn sé eðlilegt. Það veikir þessa rannsókn að sjúklingarnir voru tiltölulega fáir en rannsóknin náði þó til 14 af 15 þálifandi sjúklingum hérlendis.
Rannsóknir á fólki með einhverfu
Einhverfa (autism) er meðfætt ástand sem er mun algengara hjá drengjum en stúlkum og er í ríkum mæli arfbundið. Á síðustu árum hefur þó sannast að þekkt fósturskemmandi efni (talídómíð) getur valdið einhverfu. Ljóst er ennfremur að einhverfa er tiltölulega oft samfara skemmdum á augum, eyrum, miðtaugakerfi og fleiri líffærakerfum sem orðið hafa til snemma á meðgöngu (35-37). Nokkuð einfaldað má þess vegna líta svo á að einhverfa sé sjúkdómsmynd samfara áberandi misþroska eða þroskahefti í miðtaugakerfi. Jafnframt er fátt sem bendir til þess að í heila einhverfra sé á ungum aldri að finna hrörnunarbreytingar með próteinútfellingum í áður fullþroska taugafrumum. Fólk á ungum aldri með einhverfu ætti þannig að vera vel fallið til samanburðar við sjúklinga sem haldnir eru fyrrgreindum hrörnunarsjúkdómum. Við töldum því fýsilegt að bera saman þéttni kopars, magn og virkni cerúlóplasmíns og virkni SOD1 í blóði sjúklinga með hrörnunarsjúkdóma í miðtaugakerfinu annars vegar og einhverfu hins vegar.
Við leituðumst við að gera fullmótaða tvenndarrannsókn á ungu fólki (18-26 ára) með einhverfu í samanburði við heilbrigða einstaklinga af sama kyni og aldri. Af einhverjum sökum vildi einhverft fólk eða aðstandendur þeirra í ótrúlega mörgum tilvikum ekki taka þátt í slíkri rannsókn, en 13 af 35 sem leitað var til gáfu samþykki til rannsóknarinnar. Niðurstöðutölur benda ekki til þess að marktækur munur sé á oxunarvirkni cerúlóplasmíns eða virkni SOD1 í blóði fólks með einhverfu annars vegar og blóði fólks í samanburðarhópnum hins vegar. Þéttni cerúlóplasmíns var meiri í blóði fólks með einhverfu en í fólki í samanburðarhópnum, en munurinn var ekki marktækur. Marktækur munur var heldur ekki á þéttni kopars (Þórsdóttir og meðhöfundar, óbirtar niðurstöður). Þessar niðurstöður renna því stoðum undir þá skoðun að minnkun eða aukinn breytileiki á virkni oxavarnarensíma tengist "virkum" sjúkdómum í miðtaugakerfi, en ekki meðfæddu eða "kyrrstæðu" ástandi, eins og einhverfu, sem einkennist af misþroska fremur en hrörnunarbreytingum í áður fullþroska taugafrumum.
Rannsóknir á sauðfé með tilliti til riðu
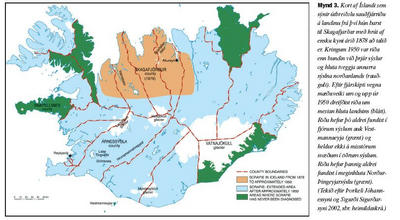
Riða er talin hafa borist til Íslands seint á 19. öld. Riða hefur síðan borist víða um landið og hefur fundist í öllum sýslum nema fjórum auk Vestmannaeyja (mynd 3). Fyrst um það bil 100 árum síðar hófust markvissar aðgerðir til þess að útrýma riðu með niðurskurði, fjárskiptum, sótthreinsun og margs konar þrifum og fleiru. Nýjum tilfellum af riðu hefur nú fækkað mjög hér á landi og útbreiðsla hennar hugsanlega stöðvast, enda þótt grunur leiki á að riða kraumi víða undir og gæti blossað upp á ný ef fyllsta aðgæsla er ekki viðhöfð (11).
Ein skýringartilgáta um uppkomu riðu gerir ráð fyrir því að mangan geti hrundið kopar úr hinu eðlilega príonpróteini (PrPc) í kindum og bundist því í staðinn og þar með valdið aflögun próteinsins eða hraðað breytingu eðlilegs príonpróteins í hið sjúklega príonprótein (PrPsc). Það er því hugsanlegt að lítið magn af kopar í umhverfi fjárins og í fóðri og mikið magn af mangan gæti stuðlað að því að sauðfjárriða komi upp (11). Í tilraunum með mýs sem sýktar höfðu verið með sjúklegu príonpróteini minnkaði einnig virkni SOD1 og GPO í heilanum um sama leyti og fyrstu sjúkdómseinkenna varð vart eða nokkru fyrr. Samfara þessu urðu greindar oxunarskemmdir í heilanum og jafnframt óx þéttni mangans í heila og blóði en magn kopars minnkaði að sama skapi (38, 39). Af þessu öllu þótti því nokkuð sjálfgefið að við rannsóknir á sauðfé þyrfti einnig að taka tillit til þessara þátta.
Alls voru tekin blóðsýni úr um það bil 140 ám, tveggja til fimm ára gömlum, á 14 býlum (sem næst 10 ær á hverju býli), og virkni cerúlóplasmíns, virkni SOD1 og virkni GPO ákvarðað. Sýni til ákvarðana á ensímvirkni voru tekin tvívegis, fyrst í september 2001 þegar fé var nýkomið af fjalli og svo aftur í mars 2002. Ærnar höfðu þá verið á húsi í liðlega þrjá mánuði og voru langt gengnar með. Var þetta gert vegna þess að fóðrun á húsi og meðganga getur haft áhrif á virkni oxavarnarensíma. Sýni til málmákvarðana voru hins vegar tekin í mars 2002. Ennfremur voru tekin sýni af ull til málmákvarðana og sýni úr heila kinda frá hlutaðeigandi býlum í sláturhúsum þegar unnt var. Var þessi rannsóknarvinna að hluta til unnin með tilstyrk frá Fulbright stofnuninni.
Býlum var skipt í fjóra flokka: 1. flokkur: Aldrei riða eða riðulaust í meira en 40 ár (6 bæir; 3 bæir á Snæfellsnesi, 3 bæir í Svarfaðardal). 2. flokkur: Riðulaust í minnst 8-10 ár (5 bæir; 3 bæir í Þingvallasveit og Grímsnesi; 2 bæir í Svarfaðardal). 3. flokkur: Riðulaust í 10-15 ár, en grunur um riðu vegna uppkomu riðu á næsta bæ í janúar 2001 (2 bæir í Hrunamannahreppi). 3a flokkur: Riðubær; riða kom upp í janúar 2002 (bær í Vatnsdal), en PrPsc var staðfest í heila úr tveimur ám frá þeim bæ sem voru með klínísk einkenni. Allar ærnar sem sýni voru tekin úr töldust heilbrigðar þegar sýnin voru tekin og annað hefur ekki komið í ljós síðar (Sigurður Sigurðarson, persónulegar upplýsingar). Allt fé á riðubænum var fellt í apríl 2002.
Niðurstöðutölur úr koparákvörðunum gáfu ekki til kynna að vöntun gæti verið á kopar í íslensku sauðfé. Ákvarðanir á virkni cerúlóplasmíns sem inniheldur mörg koparatóm, bæði fyrir og í meðgöngu, bentu og eindregið til hins sama. Virkni cerúlóplasmíns var ennfremur hin sama í ám úr öllum flokkum. Mangan hefur aldrei áður verið ákvarðað í blóði í íslensku sauðfé, svo að vitað sé. Hlutfallið milli mangans og kopars (mangan/kopar) var hæst í ám í tveimur fyrstu flokkunum. Mælir þetta ásamt öðru gegn því að skortur á kopar eða ofgnótt af mangan tengist uppkomu sauðfjárriðu. Hins vegar er ekki enn fullkannað hvort aðrir málmar gætu átt hér hlut að máli.
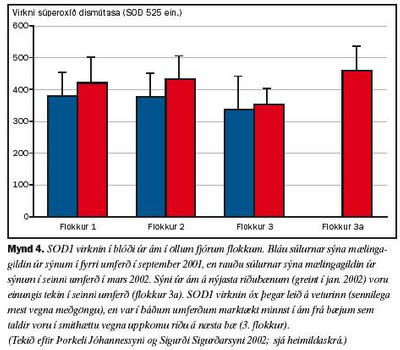
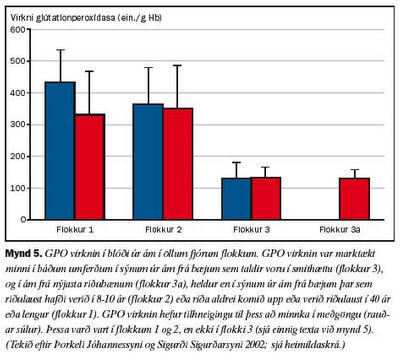
Ákvarðanir á virkni SOD1 gáfu ekki einhlíta niðurstöðu vegna þess að SOD1 virknin í ám á riðubænum (flokkur 3a) var svipuð og í ám á riðulausu bæjunum (flokkar 1 og 2), enda þótt virknin væri marktækt minni í ám á bæjum sem lágu undir grun um riðusmit (flokkur 3, sjá mynd 4). Ákvarðanir á virkni GPO bentu hins vegar til þess að ensímvirknin væri marktækt minni í ám á bæjum þar sem riða hafði komið upp eða grunur var um riðu (flokkar 3, 3a), en í ám á riðulausum bæjum (flokkar 1, 2, sjá mynd 5).
Eins og áður er nefnt tókum við sýni til ensímákvarðana tvisvar. Virkni cerúlóplasmíns var að vori marktækt meiri í ám í öllum flokkum en hafði verið haustið áður þegar fyrri sýnin voru tekin. Enginn marktækur munur var samt á virkni cerúlóplasmíns í sermi áa í mismunandi flokkum að vori fremur en að hausti svo sem áður er á drepið. Virkni SOD1 sýndi sömu tilhneigingu, en aukin virkni í meðgöngu var þó áberandi minnst í 3. flokki. Virkni GPO fór hins vegar minnkandi frá hausti til vors í ám frá bæjum í 1. og 2. flokki, en stóð í stað í ám frá bæjum í 3. flokki þar sem grunur var um riðu. Hvort sem litið er á virkni SOD1 að hausti eða vori er ljóst að ærnar í 3. flokki skáru sig úr. Sama átti við um virkni GPO í þessum ám. Að vori var virkni GPO í ám í flokkum 3 og 3a ennfremur nær hin sama. Fyllri greinargerð um þessar rannsóknir bíður birtingar (Barash og meðhöfundar).
Samandregið gefa þessar rannsóknir því ekki til kynna að vöntun á kopar gæti legið að baki uppkomu riðu, né virðist heldur skortur á selen vera að baki minni GPO virkni í ám í flokkum 3 og 3a (óbirtar athuganir). Minnkandi SOD1 virkni, en einkum minni GPO virkni, í blóði sauðfjár gæti samt tengst uppkomu eða líkum á uppkomu riðu. Mun fyllri rannsókna þarf þó við til að kanna þetta nánar.
Umræða, ályktanir og framtíðarhorfur
Við suma hrörnunarsjúkdóma í miðtaugakerfinu, svo sem Huntingtons sjúkdóm og nokkur fátíð afbrigði Alzheimer sjúkdóms, eru breytingar á ákvarðandi genum bein sjúkdómsorsök. Við langflesta hrörnunarsjúkdóma í miðtaugakerfinu hefur þó til þessa ekki tekist að sýna fram á breytingar á ákvarðandi genum nema í um það bil 10% tilvika í hæsta lagi. Í um það bil 90% tilvika verða þessir sjúkdómar því ekki tengdir þekktum genabreytingum. Hvort sem hrörnunarsjúkdómar í miðtaugakerfinu byrja á ungum aldri eða síðar, ganga hægt eða hratt eða tengjast þekktum genabreytingum eða ekki, liggur langoftast að baki ferli sem leiðir til samsöfnunar og útfellingar á starfrænum próteinum í óstarfhæfan massa í heilavefnum. Allar líkur eru og á því að forstigsbreytingar á starfrænum próteinum í miðtaugakerfi, sem eru undanfari samsöfnunar og útfellingar þeirra í óstarfrænan massa, beri með sér myndun súrefnisfríhópa. Ef þessi kenning er rétt ættu truflanir í oxavörnum að geta valdið miklu um uppkomu þessara sjúkdóma. Spurningin sem vaknar er því sú hvort vísbendingar eru í þá veru að marktækar truflanir væru jafnframt í starfi oxavarnarensíma í blóði sjúklinga með hrörnunarsjúkdóma í miðtaugakerfinu, er gagnast gætu við sjúkdómsgreiningu eða ef til vill mætti taka mið af við þróun lyfja.
Þær rannsóknir sem hér eru raktar benda til þess að truflanir á virkni oxavarnarensíma séu til staðar í blóði sjúklinga með alla þá hrörnunarsjúkdóma sem rannsakaðir hafa verið. Slíkar breytingar urðu hins vegar ekki greindar í blóði fólks með einhverfu, sem ekki verður talin til hrörnunarsjúkdóma í miðtaugakerfi. Sérlega áhugaverðar voru rannsóknir á Parkinson sjúklingum sem sýndu að ensímvirknin fór minnkandi með sjúkdómslengdinni. Endurrannsókn á þessum sjúklingum sem nú er hafin mun væntanlega varpa ljósi á gildi ákvarðana á SOD1 virkni og virkni cerúlóplasmíns fyrir mat á sjúkdómsferlinum. Fyllri rannsókn á gildi ákvarðana á GPO virkni í blóði sauðfjár mun og að líkindum skýra betur hvert sé forspárgildi slíkra ákvarðana um uppkomu riðu eða hættu á riðusmiti.
Ef rétt reynist að ákvarðanir á oxavarnarensímum í blóði hafi gildi við greiningu á hrörnunarsjúkdómum í miðtaugakerfinu, er líklegt að þessir sjúkdómar séu ekki eingöngu bundnir við miðtaugakerfið. Þótt sjúkdómseinkennin séu að vísu bundin við miðtaugakerfið útilokar það samt ekki að truflanir í oxavörnum mætti finna í flestum vefjum. Í þessu sambandi ber að minnast þess að oxun er mikil í miðtaugakerfinu samfara því að oxavarnir eru þar minni en í flestum öðrum líffærum. Þær rannsóknir sem hér greinir frá benda jafnframt til þess að mismunandi kunni að vera hver oxavarnarensím truflast í tilteknum sjúkdómum. Í þessu sambandi vekur sérstaka athygli að minnkandi virkni cerúlóplasmíns eða SOD1 þegar fyrir kemur verður ekki rakin til skorts á kopar, né minnkandi virkni GPO í sauðfé með grun um riðu að svo komnu máli til skorts á selen. Bendir það óneitanlega til þess að innbygging á kopar í virk sæti í ensímsameindir cerúlóplasmíns og SOD1 eða innbygging á seleni í virk sæti í GPO sé úr lagi færð. Allt þetta býður upp á fyllri rannsóknir.
Við alla hrörnunarsjúkdóma í miðtaugakerfinu, ekki síst við riðu, er að kalla mikill ofþroski eða offjölgun á svokölluðum stirnufrumum (astrocytosis) (1). Stirnufrumur eru helsta tegund sérstakra bandvefsfrumna (glíafrumur) í miðtaugakerfinu og gegna þær margvíslegu hlutverki til hjástoðar við taugafrumurnar, meðal annars við orkunám, endurupptöku boðefna og að einhverju leyti við bólgusvörun svo og við myndun örvefs, og þekkjast ekki í öðrum líffærum. Því hefur verið haldið fram að við hrörnunarsjúkdóma í miðtaugakerfinu á borð við riðu gæti frumáverkunin verið á stirnufrumurnar og það síðan orðið ákvarðandi fyrir skemmdir á taugafrumum á mismunandi stöðum í miðtaugakerfinu (40). Ef þetta er rétt gæti það þýtt að veiklaðar oxavarnir bitnuðu einungis eða fyrst og fremst á miðtaugakerfinu, enda þótt greina mætti einnig veiklaðar oxavarnir í öðrum líffærum einkennalítið.
Ensímið glútamínsýntasi sem er manganensím og eingöngu kemur fyrir í stirnufrumum er talið vera sérlega næmt gagn oxunarskemmdum. Ensím þetta skiptir meginmáli í þá veru að stýra umbrotum á glútamínsýru sem er helsta örvandi boðefnið í heilanum. Truflun á starfi glútamínsýntasa getur leitt til aukinnar glútamínsýruvirkni með eftirfarandi virkjun á margs konar ensímum og vaxandi hættu á frekari oxunarskemmdum auk breytinga í magni mangans í frumunum (31, 41). Vísbendingar eru í þá veru að virkni glútamínsýntasa sé einmitt skert í Alzheimer sjúkdómi í þeim hlutum heilans sem oftast verða verst úti í þeim sjúkdómi (42). Annað ensímkerfi sem mjög er bundið við stirnufrumur og skiptir meginmáli við orkunám í heilanum er kreatínfosfatasi. Bilun í þessu ensímkerfi virðist einnig vera áberandi í heila við Alzheimer sjúkdóm (42, 43) Lykillinn að dýpri skilningi á tilurð hrörnunarsjúkdóma í miðtaugakerfinu kann því að vera fólginn í rannsóknum á stirnufrumum engu síður en taugafrumunum sjálfum.
Ef bilaðar oxavarnir skipta umtalsverðu máli fyrir uppkomu hrörnunarsjúkdóma í miðtaugakerfinu er líklegt að um arfbundnar breytingar sé að ræða í sumum tilvikum, en í öðrum tilvikum ekki. Hér virðist þó enn vera sem næst óplægður akur til rannsókna. Hvernig sem þessu er farið og hvernig sem á er litið hlýtur tilbúningur lyfja, er efla oxavarnir líkamans samt að vera leið sem verður að skoða til fullnustu, við meðferð á hrörnunarsjúkdómum í miðtaugakerfinu. Lyf með súperoxídasavirkni (23) eða til þess að binda kopar eða zink (44) og þar með hugsanlega draga úr vefjaskemmandi verkun súrefnisfríhópa eru á tilraunastigi, hver svo sem gagnsemi þeirra til lækninga kann að verða. Eldri oxavarnandi lyf hafa sýnt einhverja virkni við Alzheimer sjúkdóm (tókóferól, östrógen) og bæði við þann sjúkdóm og Parkinson sjúkdóm (selegilín) (45, 46). Nýlegar rannsóknir benda og til þess að úbíkínón (Q10) gæti komið að gagni við Parkinson sjúkdóm (47). Önnur lítt könnuð leið til meðferðar á þessum sjúkdómum er að beita lyfjum sem binda og fjarlægja ummynduð prótein eða hindra ummyndun þeirra. Mjög nýlegar rannsóknir benda í raun til þess að slík lyf kunni að vera til eða finnast (48, 49). Vægi þessara tveggja leiða við rannsóknir á nýjum lyfjum gegn hrörnunarsjúkdómum í heila hefur og aukist vegna þess að bólusetning gegn mýildi í heilaskellum í mönnum er enn í deiglunni (50-52) og enn er óljóst hvert sé gildi salílyfja til varnar Alzheimer sjúkdómi (53, 54). Að sama brunni ber einnig sú staðreynd að lyf sem bæta eiga upp þrot eða bilun í þeim boðefnakerfum í miðtaugakerfi og í upphafi ber mest á í Alzheimer sjúkdómi (acetýlkólín) annars vegar og Parkinson sjúkdómi (dópamín) hins vegar hafa reynst ófullnægjandi, og þó sér í lagi við Alzheimer sjúkdóm (46).
Þörf á gagnlegri lyfjameðferð við þessa sjúkdóma er yfirþyrmandi í ljósi þess að við 85 ára aldur er minnst einn af hverjum þremur með Alzheimer sjúkdóm og annar hver með minnst eitt einkenni um Parkinson sjúkdóm. Aðrir hrörnunarsjúkdómar í miðtaugakerfinu eru að vísu mun fátíðari. Þessar staðreyndir vógu greinilega þungt í huga Stanley B. Prusiner, brautryðjanda í príonrannsóknum og Nóbelsverðlaunahafa, er hann setti eftirfarandi á blað: "problems caused by Alzheimer's disease and Parkinson's disease are already so great that if the prevalence of these maladies continues to increase in accordance with the changing demographic characteristics of the world population, they will bankrupt both developed and developing countries over the next 50 years" (1). Vonandi verður þessi ekki raunin. Þessi myrka spá dregur samt fram þá miklu vá sem gæti verið fyrir höndum ef ný meðferðarúrræði finnast ekki tímanlega. Hér má samt vissulega ekki gleyma því að auðveldari greining sjúkdóma býður ein sér upp á betri tök á forvörnum og ýmsum meðferðarúrræðum með eða án lyfja. Ef það ynnist hefði nokkuð miðað fram á veg.
Í Bretlandi (og víðar) tíðkaðist áður fyrr að fóðra nautgripi á mjöli unnu úr sláturúrgangi (bein, vöðvar og fleira) frá sauðfé. Þetta var fyrst bannað þar í landi árið 1988, en allmörgum árum áður hér á landi. Vaxandi líkur eru á því að upphaf kúariðu sé einmitt að rekja til þess að nautgripir hafi verið fóðraðir á mjöli unnu úr riðusmituðu sauðfé. Nú telst og sannað að hið nýja afbrigði af Creutzfeldt-Jakobs sjúkdómi megi rekja til neyslu á riðusmituðu nautakjöti. Enda þótt engar vísbendingar séu um að sauðfjárriða tengist beinlínis uppkomu Creutzfeldt-Jakobs sjúkdóms (sjá bls. 659) er nú samt sem áður talið ótvírætt af fyrrgreindum sökum að slíkt geti gerst ef nautgripir eru milliliður milli sauðfjár og manna. (Yfirlitsgrein: Transmissible spongiform encephalopathy as a zoonotic disease. ILSI Europe Report Series, 2003: 48.) Þessi sannindi gera útrýmingu á sauðfjárriðu að enn meira máli en áður.
Hjartariða (chronic wasting disease), sem nú herjar á tólf af ríkjum Bandaríkjanna og tvö af fylkjum Kanada, á líklega sömuleiðis upphaf sitt í riðusmituðu sauðfé. (Yam P. Shoot this deer. Sci Am 2003: 288; 26-31.)
Þess skal og getið að Páll A. Pálsson (1919-2003) var með fyrstu mönnum að gera ráð fyrir því samhengi riðusmitunar sem hér er lýst.
Sérstakar þakkir færum við Guðlaugu Þórsdóttur, lækni, fyrir margra ára samstarf. Hún hefur fengið samþykkt til doktorsnáms við Háskóla Íslands rannsókn á truflun á virkni cerúlóplasmíns og súperoxíðdismútasa (SOD1) í Parkinsonsjúkdómi og öðrum hrörnunarsjúkdómum í miðtaugakerfi. Þá viljum við þakka læknunum Grétari Guðmundssyni, Sigurlaugu Sveinbjörnsdóttur og Stefáni Hreiðarssyni fyrir gott samstarf. Tveir okkar (ÞJ og JK) færa þakkir Halldóri Runólfssyni, yfirdýralækni, Sigurði Sigurðarsyni, dýralækni, og Jed Barash, bandarískum læknastúdent og Fulbright-styrkþega, fyrir samstarf við riðurannsóknir í sauðfé. Þakkir eru ennfremur færðar Minningarsjóði Helgu Jónsdóttur og Sigurliða Kristjánssonar, Vísindasjóði Háskólans og Guðna Ágústssyni, landbúnaðarráðherra, en án framlaga þeirra hefðu þessar rannsóknir ekki verið unnar. Jóhönnu Edwald og Sigríði Ísafold Håkansson, Rannsóknastofu í lyfja- og eiturefnafræði, færum við kærar þakkir fyrir aðstoð við handritsgerð.
1. Prusiner SB. Shattuck lecture - Neurodegenerative diseases and prions. N Engl J Med 2001; 344: 1516-26.
2. Burns A, Byrne EJ, Maurer K. Alzheimer´s disease. Lancet 2002; 360: 163-5.
3. Bayer AJ. Molecular basis and genetic determinants of Alzheimer´s disease. Rev Clin Gerontol 1999; 9: 297-304.
4. Prospero N, Tagle D. Normal and mutant huntingtin: Partners in crime? Nat Med 2000; 6: 1208-9.
5. Cattaneo E, Rigamonti D, Zuccato C. The enigma of Huntington´s disease. Sci Am 2002; 287: 92-7.
6. Snædal J, Kristinsson J, Gunnarsdóttir S, Ólafsdóttir Á, Baldvinsson M, Jóhannesson Þ. Copper, ceruloplasmin and superoxide dismutase in patients with Alzheimer´s disease. Dement Geriatr Cogn Disord 1998; 9: 239-42.
7. Sveinbjörnsdóttir S, Hicks AA, Jónsson Þ, Pétursson H, Guðmundsson G, Frigge ML, et al. Familial aggregation of Parkinson's disease in Iceland. N Engl J Med 2000; 343: 1765-70.
8. Jóhannesson Þ. Alzheimer sjúkdómur: Meingerð og meðferðarmöguleikar. Heilbrigðismál 1998; (3): 27-9.
9. Jóhannesson Þ. Lyfjafræði miðtaugakerfisins. Menntamálaráðuneytið/Háskóli Íslands 1984. Kafli XI: 81-92.
10. Sigurðarson S. Epidemiology of scrapie in Iceland and experience with control measures. In: Sub-acute spongiform encephalopathies. Proceedings seminar CEC agricultural research programme. Eds: Bradely R, Saver M, Marchant B. Kulwer Acad Publ 1991: 233-42.
11. Jóhannesson Þ, Sigurðarson S. Sauðfjárriða - kopar, mangan og oxavarnarensím í íslensku sauðfé. Freyr 2002; 98: 56-9.
12. Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science 2002; 296: 1991-5.
13. Miller RJ, Wilson SM. Neurological disease: UPS stops delivering! Trends Pharmacol Sci 2003; 24: 18-23.
14. Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, et al. Elevated b-secretase expression and enzymatic activity detected in sporadic Alzheimer's disease. Nature Med 2003; 9: 3-4.
15. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 2002;297: 353-6.
16. McKeith IG, Perry EK, Perry RH. Report of the second dementia with Lewy body international workshop. Diagnosis and treatment. Neurology 1999; 53: 902-5.
17. Brown DR. Copper and prion disease. Brain Res Bull 2001; 55: 165-73.
18. Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002; 416: 507-11.
19. Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid b protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002; 416: 535-9.
20. Koppaka V, Axelsen PH. Accelerated accumulation of amyloid b proteins on oxidatively damaged lipid membranes. Biochemistry 2000; 39: 10011-6.
21. Borg DC. Oxygen free radicals and tissue injury. A reference outline. In: Oxygen free radicals in tissue damage. Eds. Tarr M, Sarason F. Birkhäuser, Boston 1993: Kafli 2: 12-53.
22. Markesbery WR, Carney JM. Oxidative alterations in Alzheimer´s disease. Brain Pathol 1999; 9: 133-46.
23. Cuzzocrea S, Riley DP, Caputi AP, Salvemini D. Antioxidant therapy: A new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Rev 2001; 53: 135-59.
24. Bush AI, Tanzi RE. The galvanization of b-amyloid in Alzheimer´s disease. Proc Natl Acad Sci 2002; 99: 7317-9.
25. Þórsdóttir G, Kristinsson J, Sveinbjörnsdóttir S, Snædal J, Jóhannesson Þ. Copper, ceruloplasmin, superoxide dismutase and iron parameters in Parkinson´s disease. Pharmacol Toxicol 1999; 85:239-43.
26. Þórsdóttir G, Kristinsson J, Hreiðarsson S, Snædal J, Jóhannesson Þ. Copper, ceruloplasmin and superoxide dismutase (SOD) in patients with Down's syndrome. Pharmacol Toxicol 2001; 89: 320-5.
27. Barber EF, Cousins RJ. Interleukin-1 - Stimulated induction of ceruloplasmin synthesis in normal and copper-deficient rats. J Nutr 1988; 118: 375-81.
28. Marcus DL, Thomas C, Rodriguez C, Simberkoff K, Tsai JS, Strafaci JA, et al. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer´s disease. Exper Neurol 1998; 150: 40-4.
29. Nagy Z. Mechanism of neuronal death in Down´s syndrome. J Neural Transm 1999; 57: 233-45.
30. Przedborski S, Kostic V, Jackson-Lewis V, Naini AB, Simonetti S, Fahn S, et al. Transgenic mice with increased Cu/Zn-superoxide dismutase activity are resistant to N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. J Neuroscience 1992; 12: 1658-67.
31. Coyle JT, Puttfarcken P. Oxidative stress, glutamate and neurodegenerative disorders. Science 1993; 262: 689-95.
32. Hirsch EC, Hunot S. Nitric oxide, glial cells and neuronal degeneration in parkinsonism. Trends Pharmacol Sci 2000; 21: 163-5.
33. Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med 2001; 344: 1688-1700.
34. Þórsdóttir G, Kristinsson J, Guðmundsson G, Snædal J, Jóhannesson Þ. Copper, ceruloplasmin and superoxide dismutase in amyotrophic lateral sclerosis. Pharm Toxicol 2000; 87: 126-30.
35. Trottier G, Srivastave L, Walker C-D. Etiology of infantile autism: a review of recent advances in genetic and neurobiological research. J Psych Neuroscience 1999; 24: 103-15.
36. Rodier PM. The early origins of autism. Sci Am 2000; 282: 38-45.
37. Lauritsen MB, Mors O, Mortensen PB, Ewald H. Medical disorders among inpatients with autism in Denmark according to ICD-8: A nationwide register- based study. J Autism Developm Disord 2002; 32: 115-9.
38. Wong B-S, Brown DR, Pan T, Whiteman M, Liu T, Xiadong B, et al. Oxidative impairment in scrapie-infected mice is associated with brain metals perturbations and altered antioxidant activities. J Neurochem 2001; 79: 689-701.
39. Thackray AM, Knight R, Haswell SJ, Bujdoso R, Brown DR. Metal imbalance and compromised antioxidant function are early changes in prion disease. Biochem J 2002; 362: 253-8.
40. Pattison IH, Jones KM. The astrocytic reaction in experimental scrapie in the rat. Res Vet Sci 1967; 8: 160-5.
41. Aschner M, Vrana KE, Zheng W. Manganese uptake and distribution in the central nervous system. Neuro Toxicology 1999; 20: 173-80.
42. Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, et al. Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. J Neurochem 1995; 65: 2146-56.
43. Burbeva GS, Aksenova MV, Makarenko IG. Decreased level of creatine kinase BB in the frontal cortex of Alzheimer patients. Dementia 1992; 3: 91-4.
44. Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, et al. Treatment with a copper-zink chelator markedly and rapidly inhibits b-amyloid accumulation in Alzheimer´s disease transgenic mice. Neuron 2001; 30: 665-76.
45. Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, et al. A controlled trial of selegiline, alpha tocopherol, or both as treatment for Alzheimer´s disease. N Engl J Med 1997; 336: 1216-22.
46. Palmer AM. Pharmacotherapy for Alzheimer´s disease: progress and prospects. Trends Pharmacol Sci 2002; 23: 426-32.
47. Frankish H. Coenzyme Q10 could slow functional decline in Parkinson's disease. Lancet 2002; 360: 1227.
48. Iversen L. Small drugs lead the attack. Nature 2002; 417: 231-3.
49. De Strooper B, Woodgett. Mental plaque removal. Nature 2003; 423: 392-3.
50. Lawrence DM. Promising developments in Alzheimer's immunotherapy. Lancet 2002; 360: 1227.
51. Haas C. New hope for Alzheimer´s disease vaccine. Nature Med 2002; 8: 1195-6.
52. Check E. Battle of the mind. Nature 2003; 422: 370-2.
53. Pasinetti GM, Pompl PM. Cyclo-oxygenase inhibitors and Alzheimer's: are we well ADAPTed? Lancet Neurol 2002; 1: 403-4.
54. Launer L. Nonsteroidal anti-inflammatory drug use and the risk for Alzheimer's disease. Drugs 2003; 63: 731-9.