
Fræðigreinar

Lungnaslagæðaháþrýstingur. Nýjungar í meðferð og sjúkratilfelli
Ágrip
Lungnaslagæðaháþrýstingur er sjaldgæfur en getur valdið áþján og dauða. Undanfarin ár hefur komið fram aukin þekking á meingerð og gangi sjúkdómsins, betri flokkun og ný lyfjameðferð. Nýju lyfin eru dýr en geta bætt verulega lífsgæði og lífslíkur. Hér er þessum atriðum lýst og tvö sjúkratilfelli kynnt.English Summary |
| Guðmundsson G, Gottskálksson G, Gíslason Þ Pulmonary arterial hypertension: new treatments and case report Læknablaðið 2003; 89: 959-61 Pulmonary arterial hypertension is a rare disease with substantial morbidity and mortality. In the last few years significant progress has been made in the understanding of the pathogenesis and course of the disease. New classification and drug treatment have emerged. The new drugs are expensive but can improve quality of life significantly. Given here is a brief review and two cases presented. Keywords: pulmonary arterial hypertension, treatment, case report. Correspondence: Gunnar Guðmundsson, ggudmund@landspitali.is |
Inngangur
Lungnaslagæðaháþrýstingur (e. pulmonary artery hypertension) er skilgreindur sem meðalþrýstingur (mean pulmonary artery pressure) í lungnaslagæð hærri en 25 mmHg í hvíld eða 30 mmHg við áreynslu. Önnur skilgreining sem einnig er viðurkennd er lungnaslagæðaþrýstingur í slagbili hærri en 40 mmHg sem samsvarar þríblöðkulokuleka hraða á Dopplerrannsókn sem er 3,0-3,5 m/sek. (1) Lungnaslagæðaháþrýstingur er sjaldgæfur en illvígur sjúkdómur og meðferðarmöguleikar hafa verið fáir til þessa (2). Árið 1998 hélt alþjóðaheilbrigðismálastofnunin ráðstefnu um sjúkdóminn (1). Þar var meðal annars kynnt ný flokkun á honum sem er sýnd í töflu I. Þannig er sjúkdómnum skipt eftir því hvort orsökin er þekkt eða óþekkt. Lungnaslagæðaháþrýstingur af óþekktri orsök er sjaldgæfur sjúkdómur með nýgengi 1-2 á milljón íbúa á ári. Hann er algengari hjá konum en körlum og kemur oft fram á yngri fullorðinsárum (3). Slíkum tilfellum hefur verið lýst hér á landi (4). Meðal þekktra orsaka lungnaháþrýstings eru lungnarek, langvinnur lungnateppusjúkdómur, kæfisvefn og margt fleira (1). Undanfarin ár hefur aukist skilningur á meingerð sjúkdómsins. Þannig er í dag talið að lungnaháþrýstingur sé fremur æðafrumuaukandi sjúkdómur en æðaþrengjandi (5). Óeðlileg fjölgun á æðaþelsfrumum sem og sléttum vöðvafrumum á sér stað. Þetta leiðir til þrenginga og lokana á æðum (5). Minnkuð framleiðsla er á köfnunarefnisoxíði og prostasýklíni frá æðaþelsfrumum (6). Aukning verður á endóþelíni sem er mjög kröftugt æðaherpandi efni og hvetur einnig til frumuskiptinga (7). Í fjölskyldum með lungnaháþrýsting af óþekktum orsökum hafa fundist stökkbreytingar í beinerfðamyndandi próteinviðtaka (bone morphogenetic protein receptor) gerð II sem er á litningi 2q.33. Þetta viðtæki er hluti af fjölskyldu frumuhvetjandi boðefna (8-10).
Áður fyrr voru meðferðarmöguleikar takmarkaðir og lifitími eftir greiningu stuttur, sérstaklega hjá þeim sem höfðu lungnaháþrýsting af óþekktri orsök (1). Litið var á lungnaháþrýsting sem sjúkdóm sem orsakaðist af æðaþrengingum og meðferð var í samræmi við það. Gefin voru æðavíkkandi lyf eins og kalkgangahemlar en árangur var óverulegur (11). Síðar var farið að nota önnur æðavíkkandi lyf eins og epóprótenól (prostasýklín) í æð (12). Þótt árangurinn væri betri en af öðrum æðavíkkandi lyfjum var þetta mjög kostnaðarsöm meðferð, með stöðugri inngjöf í æð með dælu og miklum hjáverkunum (12). Önnur meðferðarúrræði hafa lengi verið blóðþynning með warfaríni, súrefnisgjöf að nóttu eða allan sólarhringinn eftir þörf og hjartabilunarmeðferð með digoxíni og þvagræsilyfjum. Eftir að framkvæmd hefur verið prófun til að kanna árangur æðavíkkandi lyfja hefur verið hafin meðferð með kalkgangahemlum eða prostasýklíni í æð (2). Samhliða auknum skilningi á meingerð sjúkdómsins hafa komið fram ný lyf til meðferðar. Má þar nefna síldenafíl sem hamlar 5. og 6. gerð af fosfódíesterasa (13). Annað lyf sem hefur komið á sjónarsviðið nýlega er bósentan sem hamlar viðtæki fyrir endóþelín (14, 15). Kostur við þessi tvö lyf er sá að hægt er að að taka þau um munn. Ókosturinn við síldanafíl er sá að lyfið hefur ekki verið viðurkennt fyrir þessa ábendingu og Tryggingastofnun ríkisins hefur því ekki tekið þátt í kostnaði. Aukaverkun bósentan eru áhrif á lifrarstarfsemi og getur stundum þurft að hætta notkun lyfsins vegna þessa (15).
Sjúkratilfelli I
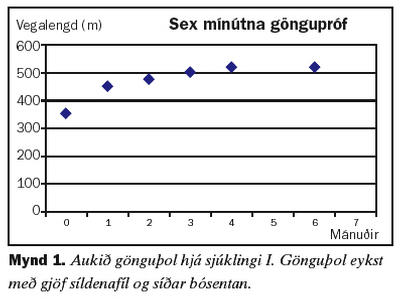
Fyrri sjúklingurinn er karlmaður sem greindist með lungnaháþrýsting árið 1998, þá 71 árs að aldri. Hafði þá haft vaxandi áreynslumæði um nokkurra ára skeið. Lungnaæðamyndatökur bentu til fyrra segareks en sjúklingur greindist einnig með kæfisvefn og hóf meðferð með ytri öndunarvél að nóttu með mismunandi þrýstingi í inn- og útöndun. Mæði var af NYHA (New York Heart Association) flokki III. Reynd var meðferð með kalkgangahemli sem þoldist illa og þrýstingur lækkaði lítið. Súrefnismeðferð var einnig hafin. Þrýstingur í lungnablóðrás fór hækkandi, mæði jókst og lífsgæði héldu áfram að skerðast. Meðalþrýstingur í lungnablóðrás var 35 mmHg. Gerð var prófun á áhrifum lyfja á þrýstinginn í lungnablóðrásinni og þrýstingur lækkaði ekki þegar hann andaði að sér köfnunarefnisoxíði (nitric oxide, NO). Hins vegar kom fram marktæk lækkun á þrýstingi eftir gjöf 25 mg af síldenafíl um munn. Hafin var meðferð með síldenafíl um munn og skammtur aukinn í 50 mg þrisvar á dag. Tryggingastofnun tók ekki þátt í lyfjakostnaði en Pfizer á Íslandi útvegaði síldenafíl sjúklingi að kostnaðarlausu. Nokkur árangur náðist, en hann var enn mæðinn og með skert gönguþol. Síðan er reynd meðferð með bósentan um munn með góðum áhrifum á lungnaþrýsting, mæði og göngugetu. Mynd 1 sýnir hvernig gönguþol jókst með lyfjagjöf. Fram kom væg hækkun á lifrarensímum sem gekk yfir án breytinga á lyfjaskömmtun.Sjúkratilfelli II
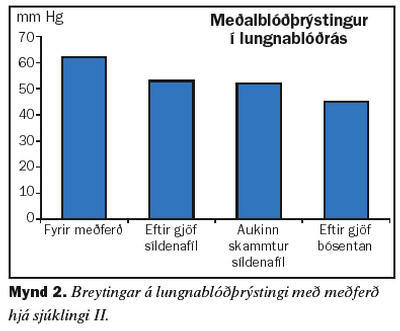
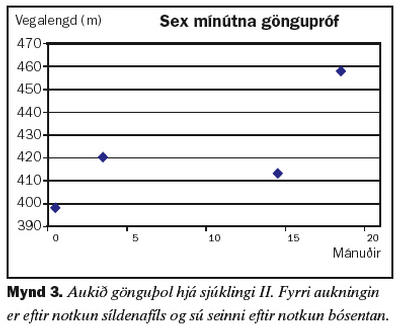
Seinni sjúklingurinn er 60 ára gömul kona sem kom til skoðunar á Lungnadeild Landspítala vegna lungnaháþrýstings árið 2001. Hún hafði verið greind með lungnaháþrýsting af völdum segareks og meðferð hafin með æðavíkkandi lyfinu hýdralazín auk blóðþynningar með warfaríni. Við eftirlit kom í ljós að meðalþrýstingur í lungnablóðrásinni var 62 mmHg. Tölvusneiðmynd af lungum sýndi ekki fram á blóðsega í miðlægum lungnaslagæðum. Svefnrannsókn greindi ekki kæfisvefn en hún var með lága súrefnismettun í svefni. Mæði var af NYHA flokki III. Gerð var prófun á áhrifum lyfja á þrýstinginn í lungnablóðrásinni og hún svaraði ekki epópróstenól í stigvaxandi skömmtum. Hins vegar kom fram marktæk lækkun á þrýstingi eftir gjöf 25 mg af síldenafíl um munn. Hafin var meðferð með síldenafíl 25 mg um munn þrisvar á dag og súrefni að næturlagi. Við eftirlit þremur mánuðum síðar var mæði minni og þrýstingur lægri. Sex mánuðum seinna var þrýstingurinn hækkandi og síldenafíl var aukið í 50 mg þrisvar á dag um munn. Við eftirlit níu mánuðum seinna hafði lungnaslagæðaþrýstingur hækkað aftur og hún hafði vaxandi áreynslumæði og skerta göngugetu við sex mínútna göngupróf. Var þá gerð lyfjaprófun með bósentan og lækkað þrýstingur marktækt við gjöf á 62,5 mg um munn. Hafin var meðferð með bósentan tvisvar á dag og haldið áfram með síldanafíl 50 mg þrisvar á dag. Mynd 2 sýnir breytingar á meðalþrýstingi í lungnablóðrás við meðferð. Skammtur bósentan var síðan aukinn í 125 mg tvisvar á dag. Mæði minnkaði aftur og göngugeta jókst eins og sést á mynd 3.Umræða
Báðir þeir sjúklingar sem hér er lýst voru í fyrstu meðhöndlaðir með æðavíkkandi lyfjum um munn með takmörkuðum árangri þannig að þrýstingur var hár og lífsgæði skert. Miklar framfarir hafa orðið í meðferð lungnaháþrýstings á síðustu árum (5). Með auknum skilningi á meingerð lungnaháþrýstings hafa ný lyf verið þróuð sem hafa öfluga verkan, minni hjáverkanir og auðveldara er að gefa en áður (5, 15). Hins vegar eru þessi lyf ákaflega dýr. Bósentan 125 mg tvisvar á dag kostar tæp 500 þúsund krónur á mánuði, eða tæpar sex milljónir á ári. Tryggingastofnun hefur ekki greitt fyrir síldanafíl meðferð þar sem hér er ekki um viðurkennda ábendingu að ræða. Framleiðandi lyfsins hefur látið það af hendi til sjúklinga endurgjaldslaust. Fjölþjóða rannsóknir á áhrifum síldenafíls á lungnaháþrýsting eru í gangi og mun hlutverk þess þá skýrast betur. Vandamál við fátíða alvarlega sjúkdóma er að erfitt er að afla sömu óyggjandi sannana í samanburðarrannsóknum. Ef lungnaháþrýstingur er orsakaður af blóðsegum í stórum lungnaslagæðum er hægt að gera aðgerð og fjarlægja innþel æðanna. Þessar aðgerðir hafa verið framkvæmdar á undanförnum áratug með góðum árangri á nokkrum stöðum í heiminum. Þær leiða til lækkunar lungnaæðaþrýstings og bæta lifun þessara sjúklinga verulega. Þær eru hins vegar mjög vandasamar og dánartíðni við þær er umtalsverð (16). Þekking á meingerð og meðferð lungnaslagæðaháþrýstings á enn eftir að aukast á næstu árum.Heimildir
1. Rich S. Executive summary from the World symposium on primary pulmonary hypertension 1998. www.who.int/ncd/cvd/ pph.htm2. Gibbs JSR, Higenbottam TW. Recommendations on the management of pulmonary hypertension in clinical practice. Heart 2001; 86: i1-i13.
3. Runo, JR, Loyd, JE. Primary pulmonary hypertension. Lancet 2003; 361: 1533-7.
4. Gíslason I, Jónasson F, Stefánsson E. Exudative retinal detachment in familial pulmonary hypertension. Acta Ophthalmol 1991; 69: 805-9.
5. Hoeper MM, Nazzareno G, Simonneau G, Rubin LJ. New treatments for pulmonary arterial hypertension. Am J Respir Crit Care Med 2002; 165: 1209-16.
6. Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 1995; 333: 214-8.
7. Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med 1993; 328: 1732-9.
8. Nichols WC, Koller DL, Slovis B. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31-32. Nat Gen 1997; 15: 277-80.
9. Deng Z, Haghighi F, Helleby L, Vanterpool K, Horn EM, Barst RJ, et al. Fine mapping of PPH1, a gene for familial primary pulmonary hypertension, to a 3-cM region on chromosome 2q33. Am J Respir Crit Care Med 2000; 161: 1055-9.
10. Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, et al. Familial primary pulmonary hypertension (Gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 2000; 67: 737-44.
11. Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992; 327: 76-81.
12. Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Herve P, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002; 40: 780-8.
13. Michelakis E, Tymchak W, Lien D, Webster L, Hashimoto K, Archer S. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation 2002; 105: 2398-403.
14. Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson, VF et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet 2001; 358: 1119-23.
15. Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002; 346: 896-903.
16. Fedullo PF, Auger WR, Kerr KM, Rubin LJ. Chronic thromboembolic pulmonary hypertension. N Engl J Med 2001; 345: 1465-72.