
Fræðigreinar
Meðfæddur IgA skortur
Ágrip
IgA skortur er einn algengasti meðfæddi ónæmisgallinn og ræðst algengi hans meðal annars af kynþætti og þjóðerni. Hjá þjóðum N-Evrópu er algengið á bilinu 1/400-1/700. Einnig er þekkt að aðrir ónæmisgallar eins og IgG2 skortur og Louis-Bar heilkenni (ataxia telangiectasia) finnist hjá einstaklingum með IgA skort. IgA finnst í hvað mestum mæli á yfirborði slímhúðarinnar. Því er athyglivert að IgA skortur eykur líkur einstaklinga á að fá sjúkdóma er herja einna helst á slímhúðina og má þar nefna endurteknar sýkingar, ofnæmi og sjálfsofnæmissjúkdóma. Auk þess virðist þessum einstaklingum hættara við að fá krabbamein. Komið hefur í ljós að í sumum tilvikum hefur IgA skortur legið í ættum. Ættfræðilegar rannsóknir hafa þannig leitt í ljós tengsl við genasvæði á litningum 6, 14, 18 og 22. Auk þess virðast ákveðnar HLA samsætur hafa sterk tengsl við sjúkdóminn. Þrátt fyrir ítarlegar rannsóknir á orsökum og afleiðingum IgA skorts er mörgum lykilspurningum enn ósvarað og þá sérstaklega hvaða aðrir hugsanlegir virkir áhættuþættir leiða til ofangreindra sjúkdóma.English Summary |
| Jörgensen GH, Lúðvíksson BR The current concept of primary IgA deficiency and its prevalence in Iceland Læknablaðið 2001; 87: 873-9 IgA deficiency is among the most common primary immune deficiency known. Its prevalence, ranging from 1/324-1/1850, depends upon the study group geographic location and its ethnicity. IgA deficiency is commonly associated with other immune defects such as IgG2, and IgG4 deficiency. In addition, ataxia telangiectasia has been associated with IgA deficiency as well. The clinical significans of IgA deficiency is presently unclear. However, increased susceptibility to atopy, autoimmunity, infections and cancer has been reported. Furthermore, majority of these diseases are bound to the mucosal surfaces; the organ where IgA is thought to have its most protective role. Recent studies focusing on the genealogy of primary IgA deficiency have found linkages to chromosome 6, 14, 18 and 22. In addition, a link to certain HLA haplotypes has been reported. Thus, further studies into the immunogenetics of IgA deficiency are needed, particularly focusing upon the question why some individuals with IgA deficiency are prone to diseases whereas others are not. In this article some of these questions are addressed, and the current literature on the topic reviewed. Correspondence: Björn Rúnar Lúðvíksson. E-mail: bjornlud@landspitali.is Key words: IgA deficiency, prevalence, immunogenetics. |
Inngangur
IgA mótefnaskortur er einn algengasti meðfæddi ónæmisgallinn og er skilgreindur sem styrkur í sermi undir 0,05 g/L. Algengið er 1/324-1/1850 og ræðst meðal annars af kynþætti og þjóðerni og hefur það reynst vera 1:633 hjá íslenskum blóðgjöfum (1). Þrátt fyrir umtalsverða vitneskju um helstu sjúkdóma sem eru samfara skortseinkennum (sýkingar, ofnæmi og sjálfsofnæmi) þá er enn óljóst af hverju einungis sumir fá slík einkenni samfara IgA skorti en aðrir ekki. Auk þess sem algengi slíkra einkenna við IgA skort er óljóst (2-13). Þrátt fyrir umtalsverðar rannsóknir á erfðamynstri IgA skorts hefur ekki fundist viðhlítandi erfðafræðileg skýring á sjúkdómnum. Á undanförnum misserum hefur skilningur manna á stjórn IgA myndunar aukist og hafa rannsóknir leitt í ljós að til að IgA framleiðsla geti hafist þarf náið samspil B- og T-eitilfrumna að eiga sér stað. Ennfremur er ljóst að til þess að slíkt samstarf geti leitt til IgA myndunar þarf einnig að koma til seyting á TGF-ß og virkjun í gegnum CD40 á B-frumum og CD40L á T-frumum (8,14-20). Auk þess hafa báðar þessar boðleiðir verið tengdar tilurð ofangreindra sjúkdóma. Í þessari yfirlitsgrein er staða þekkingar á IgA skorti í dag rædd og að hvaða þáttum frekari rannsóknir munu beinast.
Bygging IgA
IgA er 160 kDa prótein sem er samsett af tveimur sams konar léttum keðjum, kappa (k, litningur 2) eða lambda (l, litningur 22) og tveimur þungum a keðjum (litningur 14) sem IgA dregur nafn sitt af. IgA skiptist svo í tvo undirflokka (isotypes), IgA1 og IgA2, vegna ólíkra CH gena (constant region) a keðjunnar á litningi 14 (21).Dreifing IgA
IgA finnst bæði í sermi og í slímhúðarseyti. Flutningur IgA milli þessara tveggja kerfa er lítill en þau tengjast að því leyti að ef einstakling skortir IgA í sermi þá skortir hann það einnig í slímhúðarseyti. Hið gagnstæða þarf ekki að eiga við (sjá síðar). IgA getur bæði verið einliða (monomeric) og tvíliða (dimeric). IgA í sermi er 85% einliða en 15% tvíliða og 90-95% er af undirflokki IgA1 (4, 22).IgA í slímhúðarseyti er að mestu leyti tvíliða. Hlutfallsleg skipting undirflokka IgA í slímhúðarseyti er frábrugðin þeirri í sermi og hlutfall IgA2 er meira, sérstaklega eftir því sem neðar dregur í meltingarveginn (23). Líkleg skýring er sú að tvíliða IgA2 þolir vel IgA1-sértæka próteasa sem myndaðir eru af ýmsum örverum.
Myndun og útskilnaður IgA
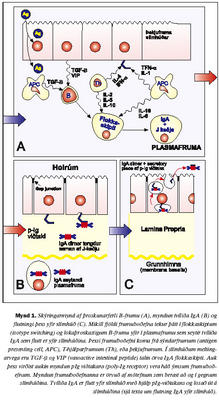
Styrkur IgA í sermi fullorðins einstaklings er 0,78-4,96 g/L (viðmiðunarmörk Rannsóknastofu Háskólans í ónæmisfræði). IgA er framleitt af plasmafrumum í beinmerg og einnig í eitlum og milta (21,22). IgA í slímhúðarseyti er jafnframt losað frá plasmafrumum sem liggja rétt undir grunnhimnu slímhúðarinnar (4). Helmingunartími IgA er þrír til sex dagar í blóði. Talið er að lýsósómal niðurbrot í lifur sé mikilvægasta niðurbrotsferli IgA í sermi en losun út í gall skipti minna máli. Hækkar því IgA í blóði meira og hraðar í starfsvefjar- (parenchymal) lifrarsjúkdómum heldur en gallvegasjúkdómum (22,24,25). Seytun IgA út á slímhúð: Meirihluti IgA í slímhúðarseyti er á tvíliða formi, tengt saman af próteinkeðju (J keðja; litningur 4). Seytun IgA er ekki eingöngu háð losun úr plasmafrumum, þar sem þekjufrumur slímhúðar hafa á basolateral helmingi viðtaka sem kallast poly-Ig viðtaki (litningur 1) sem tengist tvíliða-IgA og flytur það (transcytosis) yfir þekjuna (mynd 1). Hluti af viðtakanum (secretory component, SC) brotnar frá og eykur stöðugleika og virkni IgA á slímhúðaryfirborðinu (26-28). Galli í myndun eða starfsemi J keðju eða poly-Ig viðtaka getur því leitt til þess að sértækt IgA skorti í slímhúðarseyti þrátt fyrir að skortur sé ekki til staðar í sermi (29,30). Örlítið brot af heildarmagni IgA í slímhúðum er einliða IgA (monomeric) sem talið er flæða yfir þekjuna á milli þekjufrumna (paracellular leið) (31).
Hlutverk IgA
IgA í slímhúðarseyti er hluti af sértæka (adaptive) ónæmiskerfinu og vinnur með ósértækum vörnum slímhúðar að verndun hennar. IgA er fyrst og fremst hlutleysandi mótefni (neutralization Ig) fyrir hættuleg toxín og örverur. Auk þess hemur það viðloðun og íferð örvera og próteina í slímhúð. IgA ræsir lítið sem ekkert komplímentkerfið sem er í raun kostur þar sem slíkt myndi valda skemmdum á slímhúðinni og veikja ósértæku varnirnar (21,22,27). IgA getur einnig gert vírusa, sem staðsettir eru inni í þekjufrumunum, óvirka með því að flytja þá út í slímhúðarseytið (27). Tvíliða IgA er mun virkara en bæði einliða IgA og IgG við að gera vírusa hlutlausa. IgA í slímhúðarseyti er einnig álitið geta örvað tap á plasmíðum í bakteríum sem tjá viðloðunarprótein og sýklalyfjaónæmi (32).Hlutverk IgA í sermi er ekki jafnljóst og erfiðara að rannsaka. Einstakling sem skortir IgA í sermi skortir það einnig í slímhúðarseyti og þess vegna eru klínísku einkennin aldrei eingöngu vegna skorts í sermi. Hugsanlegt er að IgA fjarlægi lítil prótein úr blóðrásinni sem komast þangað gegnum meltingar- og öndunarveg. Þessi prótein eru þannig gerð hlutlaus áður en ofnæmis- og/eða bólgusvar myndast gegn þeim. Hefur þetta meðal annars vera talin ein af ástæðum þess að einstaklingar með IgA skort eru líklegri að fá fæðuofnæmi en þeir sem ekki eru með IgA skort (10,22).
Algengi IgA skorts
Algengi IgA skorts er mismunandi eftir kynþætti og þjóðerni. Algengi í N-Evrópu hefur mælst um einn af hverjum 600 einstaklingum (1,33-35). Í ákveðnum héruðum í Bandaríkjunum hefur algengið mælst einn af hverjum 328 einstaklingum (36), á meðan Asíuþjóðir eins og Japan hafa mælst með algengið einn af hverjum 18.500 einstaklingum (37). Algengið er oftast metið út frá mælingum á IgA í sermi blóðgjafa en árið 1964 var fyrst sýnt fram á IgA skort í heilbrigðum einstaklingum. Rannsóknum ber saman um að algengi IgA skorts meðal blóðgjafa sé á bilinu 1:400-1:700 hjá þjóðum N-Evrópu og N-Ameríku. Þar sem blóðgjafar eru fyrst og fremst heilbrigðir, ungir menn verður að teljast líklegt að algengið geti verið hærra. Íslenskar rannsóknir á IgA skorti
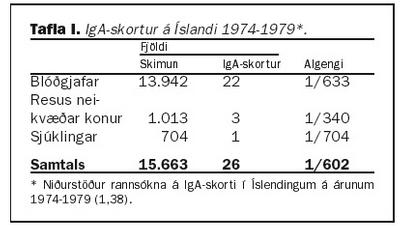
Algengi IgA skorts í heilbrigðum einstaklingum hefur áður verið rannsakað á Íslandi. Á árunum 1974-1979 var skimað fyrir IgA skorti hjá blóðgjöfum Blóðbankans (tafla I). Algengið mældist 1:633 sem til þessa hefur verið notað sem algengi IgA skorts í heilbrigðum Íslendingum. Í sömu rannsókn var skimað fyrir IgA skorti hjá 704 sjúklingum og fannst einn, algengið 1:704. Einnig var mælt IgA hjá 1.013 barnshafandi konum, sem voru resus neikvæðar og fundust þrjár, algengið 1:340 (1,38).Á árunum 1989-1992 var gerð rannsókn hér á landi þar sem 179 ungabörn á aldrinum 18-23 mánaða voru valin af handahófi og meðal annars mælt hjá þeim IgA. Þar reyndist eitt barn vera með algjöran skort (IgA undir 0,05 g/L), algengið 1:179 (39). Af þessu má sjá að mikilvægt er að meta betur algengi IgA skorts hér á landi. Ræðst það sérstaklega af því hversu lítið ungbarnaþýðið var og hversu strangar reglur eru um það hverjir mega gefa blóð. Einnig er ljóst að langflestir blóðgjafar eru karlmenn. En slíkt er mikilvægt þar sem margir sjálfsofnæmissjúkdómar eru algengari hjá konum en körlum.
Orsakir IgA skorts
IgA skortur er oftast afleiðing stöðvunar á eðlilegri sérhæfingu B-frumna yfir í IgA seytandi plasmafrumur. Þetta er flókið ferli og ekki að öllu ljóst ennþá. Meginþættir þessa ferlis eru: 1. Umbreyting forstigs B frumu (IgM+ IgD+) í eitilfrumu sem hefur IgA mótefni á yfirborði sínu (IgM+ IgD+ IgA+).
2. Þroskunarferli B frumu í IgA seytandi plasmafrumu.
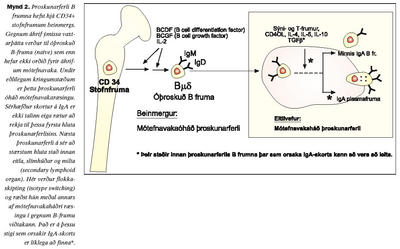
Til þess að þessum tveimur þroskastigum sé náð þarf margþætt samstarf eitilfrumna og annarra sýndarfrumna (antigen presenting cells, APC) að koma til sem felst meðal annars í ræsingu nokkurra sérhæfðra yfirborðssameinda og seytingu boðefna. Rannsóknir benda til að einstaklingar með IgA skort virðast hafa eðlilegt magn IgA yfirborðsberandi B-frumna þannig að gallinn liggur líklega í því skrefi sem kemur þar á eftir (4,14,20,40).
Fyrri rannsóknir tengdar ónæmissvari barna hafa leitt í ljós að mikilvægt er að öðlast sem skilvirkast IgA slímhúðarsvar strax á unga aldri. Íslensk rannsókn leiddi í ljós að börn sem voru með lágt IgA við 18-24 mánaða aldur voru líklegri til þess að hafa ofnæmi og þjást af eyrnabólgu (39). Jafnframt leiddu framhaldsrannsóknir það í ljós að fjögurra ára börn með lágt IgA í slímhúð voru líklegri til þess að þjást af ofnæmiskvefi og hafa jákvæða svörun á ofnæmishúðprófum. Því er verulega mikilvægt að auka skilning okkar bæði á útbreiðslu og tengslum IgA skorts við kvilla sem gætu tengst ónæmisfræðilegum vandamálum. Er þetta sérstaklega áhugavert þar sem að stjórnunarferli IgA myndunar er háð að minnsta kosti bæði seytingu á TGF-b og tjáningu á CD40-CD40L, en bæði þessi boðleiðakerfi hafa mikilvægu hlutverki að gegna í almennri bólgusvörun. Jafnframt hefur verið leitt í ljós að einstaklingar sem þjást af CD40L skorti (Hyper IgM syndrome) geta ekki myndað aðra mótefnaundirflokka en IgM (41). Flokkaskiptaferlið (isotype switching) í B-frumum er háð efnaþáttum sem losaðir eru meðal annars af APC-frumum, T-frumum og B-frumunum sjálfum. Galli eða skortur á TGF-b og IL 4-5-6-10 hefur verið athugaður sem hugsanleg orsök IgA skorts. Sýnt hefur verið fram á marktækt lægri gildi TGF-b í einstaklingum með IgA skort, en mikilvægi þess er enn óljóst (14,19,42-50). Hugsanlegir gallar í viðtökum þessara efna hafa einnig verið nefndir sem hugsanlegir meinvaldar (15,18,41,51). Af þessu má vera ljóst að líklegt verður að teljast að hjá þeim einstaklingum, þar sem IgA skortur og ónæmismiðlaðir sjúkdómar fara saman, megi búast við göllum í einhverju af ofangreindum stjórnunarferlum (mynd 2).
Erfðafræðilegir þættir IgA skorts: IgA skortur er í sumum tilvikum bundinn við fjölskyldur. Þar sem skorturinn er ekki bundinn kynjum gæti hans verið að leita í stökkbreytingu gens, með takmarkaða sýnd, sem erfist ríkjandi á A-litningi. Í dag eru nokkur genasvæði talin vera álitlegust.
1. Á litningi 6 er Major Histocompatibility Complex (MHC). IgA skortur hefur verið tengur ákveðnum HLA samsætum (haplotypes) og MHC genaafurðir eru taldar taka þátt í myndun IgA. Þar sem MHC gen af flokki II og afurðir þeirra eru nauðsynlegar starfsemi T-hjálparfrumna og tengslum þeirra við B-frumur þá er rökrétt að álykta að MHC gen af flokki II geti valdið IgA skorti. Þessu til stuðnings hefur IgA skortur verið tengdur ákveðnum genum af flokki II, einkum þeim sem innihalda HLA DR DQ samsætur (52,53). Aðrar rannsóknir hafa sýnt að MHC gen af flokki III hafi sterkari tengsl við þær samsætur sem helst tengjast IgA skorti. Gen af flokki III tjá komplímentþættina C2 og C4 og þátt B en einnig TNF-a og TNF-b sem eru nauðsynleg eðlilegu mótefnasvari. Athygli vekur sú fylgni sem er á milli IgA skorts og þess að hafa brottfall eða tvöföldun í C4a geninu (54). Svæðið milli merkigena af flokki III D821/D823 og svo HLA-B8 inniheldur 21 gen, þar með talin áðurnefnd gen af flokki III, og er þetta svæði líklega það áhugaverðasta í dag (55,56).
2. Á litningi 14 eru staðsett gen sem tjá óbreytanlega hluta (constant region, CH) þungu mótefnakeðjanna. Brottfall eða tvöföldun á þessum genum geta hindrað flokkaskipti en áhugi manna á þessu svæði, sem meginorsök IgA skorts fer dvínandi (17,57,58).
3. IgA-skortur og CVID (common variable immune deficiency) hafa lengi verið þekkt afleiðing galla á litningi 18. Brottfall á stutta arminum eða langa arminum virðist oft á tíðum valda IgA skorti. Nýjustu rannsóknir draga þó úr hugsanlegu mikilvægi þessa litnings hvað varðar meginorsök IgA skorts (59,60).
4. Brottfall á litningi 22 getur valdið DiGeorge heilkenni (algengi 1/3-4000). Lélegt mótefnasvar og IgA skortur er algengur meðal þessara einstaklinga, en ekki er vitað hvort það er vegna lélegrar T-frumna starfsemi eða vegna óþekkts B-frumna galla (61).
Áunnar orsakir IgA skorts
Mörg lyf geta valdið IgA skorti. Algengustu lyfin eru bólgueyðandi gigtarlyf og flogaveikilyf. Þannig geta súlfasalizín, gull, klórókín, karbamazepín og valpórat öll valdið IgA skorti. Í um helmingi tilvika er um afturkræfanlegan skort að ræða en hjá hinum viðhelst skorturinn þrátt fyrir að lyfjagjöf sé hætt. Ekki er vitað hvernig lyfin valda lækkuninni (62-64). Tímabundinn IgA skortur
Yfirleitt er reglan sú að ef einstaklingur greinist með IgA skort þá er skorturinn varanlegur. Tímabundinn skortur (transient deficiency) getur átt sér stað, eins og áður sagði, við vissar lyfjagjafir en einnig er honum lýst hjá börnum. Hluti barna sem greinast með IgA skort eða mjög lágt IgA virðist með tímanum geta þróað eðlilegt magn IgA þrátt fyrir að gildi þeirra liggi alltaf við neðri mörk viðmiðunarmarka. Hugsanlega liggur önnur arfgerð á bak við tímabundinn skort en varanlegan skort (39,59).Afleiðingar IgA skorts
Af þeim sjúkdómum er einstaklingar með IgA skort eru hvað útsettastir fyrir eru endurteknar sýkingar, en sjálfsofnæmissjúkdómar, ofnæmissjúkdómar og krabbamein eru einnig talin tengjast IgA skorti. Ýmsar rannsóknir hafa sýnt fram á, að stærsti hluti einstaklinga með IgA skort virðist einkennalaus. Margt bendir þó til þess að ef slíkum einstaklingum er fylgt eftir í lengri tíma, sé tíðni slímhúðarsýkinga og annarra fylgikvilla hærri (10,65). Talið var að afleiðing skorts væri háð því umhverfi sem fólk lifir í og þar með því áreiti sem slímhúðin verður fyrir en svo virðist ekki vera. Gerð var rannsókn á afleiðingum IgA skorts í fátækrahverfum Brasilíu sem sýndi að um það bil tveir þriðju einstaklinga með IgA skort voru án einkenna eða svipað hlutfall og hjá þjóðfélagshópum er búa við betri kjör (6). Hugsanlegt er að einstaklingar með skort án einkenna nái að einhverju leyti að vega upp á móti skortinum með því að auka aðra varnarþætti. Aukið magn IgM í slímhúðarseyti og aukinn styrkur IgG í sermi mælist oft hjá einstaklingum með IgA skort (1,6,66). Konur með IgA skort, sem hafa börn á brjósti, mælast með margfalda hækkun á bæði IgM og IgG í brjóstamjólk sem leiðir til þess að mjólkin er sneisafull af mótefnum (7). Skortur á IgG2 og IgG4 undirflokkum finnst hjá 20-30% einstaklinga með IgA skort og þessi undirhópur er líklegri til að sýna einkenni (2,9,65). Sýkingar: Við IgA skort er aðallega um að ræða endurteknar slímhúðarsýkingar eða lífshættulegar innri (systematic) sýkingar eins og heilahimnubólgur, blóðsýkingar eða lifrarbólgur (10). Oftast er þó um að ræða aukna tíðni sýkinga í efri og neðri loftvegum. Sýkingar í meltingarvegi geta verið bráður eða langvinnur niðurgangur (Giardia lamblia) (2,67). Sýkingar í gallvegum geta leitt til langvinnrar gallblöðrubólgu og gallsteinamyndunar. Aukinni tíðni langvinnra sveppasýkinga, bæði í húð og slímhúðum, hefur einnig verið lýst hjá einstaklingum með IgA skort (4).
Sjálfsofnæmissjúkdómar: Einstaklingar með IgA skort eru líklegri en aðrir til að mynda sjálfsmótefni gegn margs konar vefjaskyldum mótefnavökum. Líkleg ástæða fyrir því er aukin íferð ýmissa próteina og annarra mótefnavaka, inn í slímhúðir og í blóðrás, sem hefði annars verið hindruð af IgA. Aukið magn sjálfsmótefna er tengt aukinni tíðni sjálfsofnæmissjúkdóma hjá einstaklingum með IgA skort. Rauðir úlfar og iktsýki hafa fundist hjá allt að 5% einstaklinga með IgA skort og aukin tíðni er af skjaldkirtilsbólgu, sóra, dermatomyositis (vöðvaþrota í húð), acute hemolytic anemia (bráðu rauðlosablóðleysi) og Sjögrens sjúkdómi (13,40,68-70). Þessir sjálfsofnæmissjúkdómar eru taldir bein afleiðing IgA skorts en svo eru aðrir sjúkdómar sem finnast í auknu mæli hjá einstaklingum með IgA skort, en eru ekki taldir afleiðing IgA skorts heldur álitnir spretta af sama erfðafræðilega gallanum. Hinir síðarnefndu eru insúlínháð sykursýki og glútenóþol en fundist hefur ákveðin MHC samsæta sem tengd hefur verið við þessa tvo sjúkdóma og IgA skort (5,71-74).
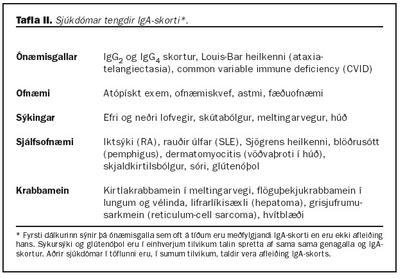
Krabbamein: Illkynja sjúkdómar hafa verið tengdir við IgA skort og þá sérstaklega kirtlakrabbamein í meltingarvegi (tafla II). Aukin íferð mótefnavaka í slímhúðir getur valdið langvarandi bólgumyndun sem eykur líkur á myndun illkynja frumna (75). Einnig hafa birst greinar um hærri tíðni hvítblæðis hjá einstaklingum með IgA skort (76).
Ofnæmissjúkdómar: Ofnæmissjúkdómar eins og ofnæmiskvef, astmi og exem finnast í auknu mæli hjá einstaklingum með IgA skort. Talið er líklegt að vanhæfni til að mynda slímhúðarmótefni ýti undir framleiðslu ofnæmis IgE mótefna gegn mótefnavökum sem við öndum að okkur eða tökum inn gegnum húð eða slímhúð (3,10,39,43,77,78).
Ofnæmislost: Meira en 20% einstaklinga með IgA skort mynda mótefni gegn IgA og hluti þeirra er í sérstakri hættu á að fá ofnæmislost gegn blóðafurðum sem innihalda IgA.
Staða þekkingar IgA skorts á Íslandi
Einu rannsóknirnar sem að vitað er til að gerðar hafi verið hérlendis eru ofangreindar rannsóknir á vegum Blóðbankans á algengi IgA skorts meðal blóðgjafa og ættartengslum þeirra og rannsókn á astma og ofnæmi meðal barna, fæddra 1987 á Landspítalanum (1,38). Af þessum rannsóknum hafa fengist veigamiklar upplýsingar sem meðal annars mun gagnast til áframhaldandi rannsókna og hafa helstu niðurstöður þeirra verið tíundaðar hér að ofan.Vegna mikilla framfara innan sameindaerfðafræði og líffræði hafa nú opnast möguleikar á að rannsaka með enn nákvæmari hætti, en þegar ofangreindar rannsóknir fóru af stað, IgA skort meðal Íslendinga. Því hefur nú verið ýtt af stað umfangsmiklu samstarfsverkefni Rannsóknastofu Háskólans í ónæmisfræði, Blóðbankans og rannsóknastofu í meinefnafræði, Landspítala Hringbraut á orsök, afleiðingu og algengi IgA skorts á meðal, annars vegar heilbrigðra blóðgjafa og hins vegar einstaklinga sem leita eftir þjónustu lækna. Meginmarkmið rannsóknarinnar er að athuga erfðafræðilega meingerð og orsök IgA skorts. Fyrstu niðurstöður rannsóknarinnar liggja nú fyrir og verða vonandi kynntar innan tíðar fyrir lesendum Læknablaðsins. Þar kemur meðal annars fram að hugsanlegt er að afleiðingar IgA skorts í dag geti verið alvarlegri en menn hafa almennt gert ráð fyrir fram að þessu.
Heimildir
1. Ulfarsson J, Gudmundsson S, Birgisdottir B, Kjeld JM, Jensson O. Selective serum IgA deficiency in Icelanders. Frequency, family studies and Ig levels. Acta Med Scand 1982; 211: 481-7.
2. Aittoniemi J, Koskinen S, Laippala P, Laine S, Miettinen A. The significance of IgG subclasses and mannan-binding lectin (MBL) for susceptibility to infection in apparently healthy adults with IgA deficiency. Clin Exp Immunol 1999; 116: 505-8.
3. Brasher GW, Bourland PD. The role of IgA in the pathogenesis of atopy. Ann Allergy 1975; 34: 137-40.
4. Burrows PD, Cooper MD. IgA deficiency. Adv Immunol 1997; 65: 245-76.
5. Cataldo F, Marino V, Bottaro G, Greco P, Ventura A. Celiac disease and selective immunoglobulin A deficiency. J Pediatr 1997; 131: 306-8.
6. Castrignano SB, Carlsson B, Carneiro-Sampaio MS, Soderstrom T, Hanson LA. IgA and IgG subclass deficiency in a poor population in a developing country. Scand J Immunol 1993; 37: 509-14.
7. Hahn-Zoric M, Carlsson B, Bjorkander J, Mellander L, Friman V, Padyukov L, et al. Variable increases of IgG and IgM antibodies in milk of IgA deficient women. Pediatr Allergy Immunol 1997; 8: 127-33.
8. Benson EB, Strober W. Regulation of IgA secretion by T cell clones derived from the human gastrointestinal tract. J Immunol 1988; 140: 1874-82.
9. Johnson ML, Keeton LG, Zhu ZB, Volanakis JE, Cooper MD, Schroeder HW, Jr. Age-related changes in serum immunoglobulins in patients with familial IgA deficiency and common variable immunodeficiency (CVID). Clin Exp Immunol 1997; 108: 477-83.
10. Koskinen S. Long-term follow-up of health in blood donors with primary selective IgA deficiency. J Clin Immunol 1996; 16: 165-70.
11. Korponay-Szabo IR, Kovacs JB, Czinner A, Goracz G, Vamos A, Szabo T, et al. High prevalence of silent celiac disease in preschool children screened with IgA/IgG antiendomysium antibodies. J Pediatr Gastroenterol Nutr 1999; 28: 26-30.
12. Rankin EC, Isenberg DA. IgA deficiency and SLE: prevalence in a clinic population and a review of the literature. Lupus 1997; 6: 390-4.
13. Steuer A, McCrea DJ, Colaco CB. Primary Sjogren's syndrome, ulcerative colitis and selective IgA deficiency. Postgrad Med J 1996; 72: 499-500.
14. Ehrhardt RO, Strober W, Harriman GR. Effect of transforming growth factor (TGF)-beta 1 on IgA isotype expression. TGF-beta 1 induces a small increase in sIgA+ B cells regardless of the method of B cell activation. J Immunol 1992; 148: 3830-6.
15. Stuber E, Strober W, Neurath M. Blocking the CD40L-CD40 interaction in vivo specifically prevents the priming of T helper 1 cells through the inhibition of interleukin 12 secretion. J Exp Med 1996; 183: 693-8.
16. Elson CO, Heck JA, Strober W. T-cell regulation of murine IgA synthesis. J Exp Med 1979; 149: 632-43.
17. Harriman GR, Bogue M, Rogers P, Finegold M, Pacheco S, Bradley A, et al. Targeted deletion of the IgA constant region in mice leads to IgA deficiency with alterations in expression of other Ig isotypes. J Immunol 1999; 162: 2521-9.
18. Hiroi T, Yanagita M, Iijima H, Iwatani K, Yoshida T, Takatsu K, et al. Deficiency of IL-5 receptor alpha-chain selectively influences the development of the common mucosal immune system independent IgA-producing B-1 cell in mucosa-associated tissues. J Immunol 1999; 162: 821-8.
19. Marconi M, Plebani A, Avanzini MA, Maccario R, Pistorio A, Duse M, et al. IL-10 and IL-4 co-operate to normalize in vitro IgA production in IgA-deficient (IgAD) patients. Clin Exp Immunol 1998; 112: 528-32.
20. Wang Z, Yunis D, Irigoyen M, Kitchens B, Bottaro A, Alt FW, et al. Discordance between IgA switching at the DNA level and IgA expression at the mRNA level in IgA-deficient patients. Clin Immunol 1999; 91: 263-70.
21. Kerr MA. The structure and function of human IgA. Biochem J 1990; 271: 285-96.
22. Conley ME, Delacroix DL. Intravascular and mucosal immunoglobulin A: two separate but related systems of immune defense? Ann Intern Med 1987; 106: 892-9.
23. Kilian M, Reinholdt J, Lomholt H, Poulsen K, Frandsen EV. Biological significance of IgA1 proteases in bacterial colonization and pathogenesis: critical evaluation of experimental evidence. Apmis 1996; 104: 321-38.
24. Calvo M, Pol A, Lu A, Ortega D, Pons M, Blasi J, et al. Cellubrevin is present in the basolateral endocytic compartment of hepatocytes and follows the transcytotic pathway after IgA internalization. J Biol Chem 2000; 275: 7910-7.
25. Delacroix DL, Elkom KB, Geubel AP, Hodgson HF, Dive C, Vaerman JP. Changes in size, subclass, and metabolic properties of serum immunoglobulin A in liver diseases and in other diseases with high serum immunoglobulin A. J Clin Invest 1983; 71: 358-67.
26. Brandtzaeg P. Molecular and cellular aspects of the secretory immunoglobulin system. Apmis 1995; 103: 1-19.
27. Brandtzaeg P, Farstad IN, Johansen FE, Morton HC, Norderhaug IN, Yamanaka T. The B-cell system of human mucosae and exocrine glands. Immunol Rev 1999; 171: 45-87.
28. Renegar KB, Jackson GD, Mestecky J. In vitro comparison of the biologic activities of monoclonal monomeric IgA, polymeric IgA, and secretory IgA. J Immunol 1998; 160: 1219-23.
29. Lycke N, Erlandsson L, Ekman L, Schon K, Leanderson T. Lack of J chain inhibits the transport of gut IgA and abrogates the development of intestinal antitoxic protection. J Immunol 1999; 163: 913-9.
30. Shimada S, Kawaguchi-Miyashita M, Kushiro A, Sato T, Nanno M, Sako T, et al. Generation of polymeric immunoglobulin receptor-deficient mouse with marked reduction of secretory IgA. J Immunol 1999; 163: 5367-73.
31. Persson CG, Erjefalt JS, Greiff L, Erjefalt I, Korsgren M, Linden M, et al. Contribution of plasma-derived molecules to mucosal immune defence, disease and repair in the airways. Scand J Immunol 1998; 47: 302-13.
32. Porter P, Linggood MA. Novel mucosal anti-microbial functions interfering with the plasmid-mediated virulence determinants of adherence and drug resistance. Ann N Y Acad Sci 1983; 409: 564-79.
33. Koistinen J. Selective IgA deficiency in blood donors. Vox Sang 1975; 29: 192-202.
34. Sampaio MM, Carbonare SB, Rozentraub RB, Mamede MN, Ribeiro MA, Porto MH, et al. [Prevalence of selective IgA deficiency in blood donors and pregnant women clinically healthy [letter]]. Rev Paul Med 1987; 105: 239-40.
35. Wells JV, McNally MP, King MA. Selective IgA deficiency in Australian blood donors. Aust N Z J Med 1980; 10: 410-3.
36. Clark JA, Callicoat PA, Brenner NA, Bradley CA, Smith DM, Jr. Selective IgA deficiency in blood donors. Am J Clin Pathol 1983; 80: 210-3.
37. Kanoh T, Mizumoto T, Yasuda N, Koya M, Ohno Y, Uchino H, et al. Selective IgA deficiency in Japanese blood donors: frequency and statistical analysis. Vox Sang 1986; 50: 81-6.
38. Gudmundsson S, Jensson O. Frequency of IgA deficiency in blood donors and Rh negative women in Iceland. Acta Pathol Microbiol Scand [C] 1977; 85: 87-9.
39. Ludviksson BR, Eiriksson TH, Ardal B, Sigfusson A, Valdimarsson H. Correlation between serum immunoglobulin A concentrations and allergic manifestations in infants. J Pediatr 1992; 121: 23-7.
40. Strober W, Sneller MC. IgA deficiency. Ann Allergy 1991; 66: 363-75.
41. Ramesh N, Seki M, Notarangelo LD, Geha RS. The hyper-IgM (HIM) syndrome. Springer Semin Immunopathol 1998; 19: 383-99.
42. Bridoux F, Badou A, Saoudi A, Bernard I, Druet E, Pasquier R, et al. Transforming growth factor beta (TGF-beta)-dependent inhibition of T helper cell 2 (Th2)-induced autoimmunity by self-major histocompatibility complex (MHC) class II-specific, regulatory CD4(+) T cell lines. J Exp Med 1997; 185: 1769-75.
43. Conley ME, Cooper MD. Genetic basis of abnormal B cell development. Curr Opin Immunol 1998; 10: 399-406.
44. Del Giudice G, Crow MK. Role of transforming growth factor beta (TGF beta) in systemic autoimmunity. Lupus 1993; 2: 213-20.
45. Horwitz DA, Gray JD, Ohtsuka K, Hirokawa M, Takahashi T. The immunoregulatory effects of NK cells: the role of TGF-beta and implications for autoimmunity. Immunol Today 1997; 18: 538-42.
46. Inobe J, Slavin AJ, Komagata Y, Chen Y, Liu L, Weiner HL. IL-4 is a differentiation factor for transforming growth factor-beta secreting Th3 cells and oral administration of IL-4 enhances oral tolerance in experimental allergic encephalomyelitis. Eur J Immunol 1998; 28: 2780-90.
47. Lio D, D'Anna C, Leone F, Curro MF, Candore G, Caruso C. Hypothesis: interleukin-5 production impairment can be a key point in the pathogenesis of the MHC-linked selective IgA deficiency. Autoimmunity 1998; 27: 185-8.
48. Muller F, Aukrust P, Nilssen DE, Froland SS. Reduced serum level of transforming growth factor-beta in patients with IgA deficiency. Clin Immunol Immunopathol 1995; 76: 203-8.
49. Piccirillo CA, Chang Y, Prud'homme GJ. TGF-beta1 somatic gene therapy prevents autoimmune disease in nonobese diabetic mice. J Immunol 1998; 161: 3950-6.
50. van Ginkel FW, Wahl SM, Kearney JF, Kweon MN, Fujihashi K, Burrows PD, et al. Partial IgA-deficiency with increased Th2-type cytokines in TGF-beta 1 knockout mice. J Immunol 1999; 16: 1951-7.
51. Zan H, Cerutti A, Dramitinos P, Schaffer A, Casali P. CD40 engagement triggers switching to IgA1 and IgA2 in human B cells through induction of endogenous TGF-beta: evidence for TGF-beta but not IL-10-dependent direct S mu-->S alpha and sequential S mu-->S gamma, S gamma-->S alpha DNA recombination. J Immunol 1998; 161: 5217-25.
52. Fiore M, Pera C, Delfino L, Scotese I, Ferrara GB, Pignata C. DNA typing of DQ and DR alleles in IgA-deficient subjects. Eur J Immunogenet 1995; 22: 403-11.
53. Reil A, Bein G, Machulla HK, Sternberg B, Seyfarth M. High-resolution DNA typing in immunoglobulin A deficiency confirms a positive association with DRB1*0301, DQB1*02 haplotypes. Tissue Antigens 1997; 50: 501-6.
54. Gerbase-Delima M, Pinto LC, Grumach A, Carneiro-Sampaio MM. HLA antigens and haplotypes in IgA-deficient Brazilian paediatric patients. Eur J Immunogenet 1998; 25: 281-5.
55. Schroeder HW, Jr., Zhu ZB, March RE, Campbell RD, Berney SM, Nedospasov SA, et al. Susceptibility locus for IgA deficiency and common variable immunodeficiency in the HLA-DR3, -B8, -A1 haplotypes. Mol Med 1998; 4: 72-86.
56. Cucca F, Zhu ZB, Khanna A, Cossu F, Congia M, Badiali M, et al. Evaluation of IgA deficiency in Sardinians indicates a susceptibility gene is encoded within the HLA class III region. Clin Exp Immunol 1998; 111: 76-80.
57. Plebani A, Carbonara AO, Bottaro A, Gallina R, Boccazzi C, Crispino P, et al. Two siblings with deficiency of IgA1, IgG2, IgG4 and IgE due to deletion of immunoglobulin heavy chain constant region genes. Year Immunol 1993; 7: 231-5.
58. Plebani A, Carbonara AO, Bottaro A, Gallina R, Boccazzi C, Crispino P, et al. Gene deletion as a cause of associated deficiency of IgA1, IgG2, IgG4 and IgE. Immunodeficiency 1993; 4(1-4): 245-8.
59. Vorechovsky I, Zetterquist H, Paganelli R, Koskinen S, Webster AD, Bjorkander J, et al. Family and linkage study of selective IgA deficiency and common variable immunodeficiency. Clin Immunol Immunopathol 1995; 77: 185-92.
60. Vorechovsky I, Blennow E, Nordenskjold M, Webster AD, Hammarstrom L. A putative susceptibility locus on chromosome 18 is not a major contributor to human selective IgA deficiency: evidence from meiotic mapping of 83 multiple-case families. J Immunol 1999; 163: 2236-42.
61. Smith CA, Driscoll DA, Emanuel BS, McDonald-McGinn DM, Zackai EH, Sullivan KE. Increased prevalence of immunoglobulin A deficiency in patients with the chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Diagn Lab Immunol 1998; 5: 415-7.
62. Murphy EA, Morris AJ, Walker E, Lee FD, Sturrock RD. Cyclosporine A induced colitis and acquired selective IgA deficiency in a patient with juvenile chronic arthritis. J Rheumatol 1993; 20: 1397-8.
63. Truedsson L, Baskin B, Pan Q, Rabbani H, Vorechovsky I, Smith CI, et al. Genetics of IgA deficiency. Apmis 1995; 103: 833-42.
64. Braconier JH. Reversible total IgA deficiency associated with phenytoin treatment. Scand J Infect Dis 1999; 31: 515-6.
65. Barlan IB, Bakir M, Tukenmez F, Basaran MM. Long-term follow-up of patients with IgG subclass or IgA deficiencies [letter]. Acta Paediatr 1995; 84: 828.
66. Mellander L, Bjorkander J, Carlsson B, Hanson LA. Secretory antibodies in IgA-deficient and immunosuppressed individuals. J Clin Immunol 1986; 6: 284-91.
67. Friman V, Adlerberth I, Connell H, Svanborg C, Hanson LA, Wold AE. Decreased expression of mannose-specific adhesins by Escherichia coli in the colonic microflora of immunoglobulin A-deficient individuals. Infect Immun 1996; 64: 2794-8.
68. Strober W, Fuss IJ, Ehrhardt RO, Neurath M, Boirivant M, Ludviksson BR. Mucosal immunoregulation and inflammatory bowel disease: new insights from murine models of inflammation TGF-beta production regulates the development of the 2,4,6-trinitrophenol-conjugated keyhole limpet hemocyanin-induced colonic inflammation in IL-2-deficient mice. Scand J Immunol 1998; 48: 453-8.
69. Taniguchi S, Kawahira K, Kaneto K, Hamada T. Juvenile pityriasis rubra pilaris with isolated IgA deficiency [letter]. Eur J Pediatr 1997; 156: 896.
70. Terzioglu E, Kokuludag A, Sin A, Kirmaz C, Yalcin M, Sebik F, et al. Selective IgA deficiency and ankylosing spondylitis Spondylarthropathy and selective IgA deficiency Ankylosing spondylitis and selective IgA deficiency [letter; comment] Selective IgA deficiency and spondyloarthropathy: a distinct disease? [see comments]. J Investig Allergol Clin Immunol 1997; 7: 619-20.
71. Gandolfi L, Pratesi R, Cordoba JC, Tauil PL, Gasparin M, Catassi C. Prevalence of celiac disease among blood donors in Brazil [see comments]. Am J Gastroenterol 2000; 95: 689-92.
72. Heneghan MA, Stevens FM, Cryan EM, Warner RH, McCarthy CF. Celiac sprue and immunodeficiency states: a 25-year review. J Clin Gastroenterol 1997; 25: 421-5.
73. Hill I, Fasano A, Schwartz R, Counts D, Glock M, Horvath K. The prevalence of celiac disease in at-risk groups of children in the United States. J Pediatr 2000; 136: 86-90.
74. Loft DE, Nwokolo CU, Ciclitira PJ. The diagnosis of gluten sensitivity and coeliac disease--the two are not mutually inclusive [comment]. Eur J Gastroenterol Hepatol 1998; 10: 911-3.
75. Filipovich AH, Mathur A, Kamat D, Kersey JH, Shapiro RS. Lymphoproliferative disorders and other tumors complicating immunodeficiencies. Immunodeficiency 1994; 5: 91-112.
76. Ott MM, Ott G, Klinker H, Trunk MJ, Katzenberger T, Muller-Hermelink HK. Abdominal T-cell non-Hodgkin's lymphoma of the gamma/delta type in a patient with selective immunoglobulin A deficiency. Am J Surg Pathol 1998; 22: 500-6.
77. Hammarstrom L, Smith CI. Immunoglobulin subclass distribution of specific antibodies in allergic patients. Prediction of the IgE inducing capacity of potential allergens. Allergy 1987; 42: 529-34.
78. van Asperen PP, Gleeson M, Kemp AS, Cripps AW, Geraghty SB, Mellis CM, et al. The relationship between atopy and salivary IgA deficiency in infancy. Clin Exp Immunol 1985; 62: 753-7.