
Fræðigreinar



Arfgeng heyrnarskerðing Leit að erfðabreytileikum í genum konnexíns 26 og POU3F4 hjá einstaklingum með meðfædda heyrnarskerðingu án tengsla við heilkenni
Ágrip
Markmið: Nýlegar erlendar rannsóknir hafa sýnt að um 10-30% af alvarlegri heyrnarskerðingu hjá nýburum eru vegna erfðabreytileika í geni konnexíns 26 (Cx26) og er Cx26 35delG langalgengastur. Erfðabreytileikar í geni POU3F4 eru algengasta orsök arfgengrar heyrnarskerðingar, sem erfist kynbundið. Markmið þessarar rannsóknar var að ákvarða hvort og hvaða erfðabreytileikar í genum Cx26 og POU3F4 valdi meðfæddri heyrnarskerðingu án tengsla við heilkenni hjá íslenskum einstaklingum.Efniviður og aðferðir: Þátttakendur voru 15 einstaklingar, sem uppfylltu þau skilyrði að hafa verulega meðfædda heyrnarskerðingu af óþekktum toga, og að heyrnarskerðingin hafði verið staðfest með heyrnarmælingu. Ellefu einstaklingar höfðu fjölskyldusögu, en fjögur voru stök tilfelli. Allar útraðir Cx26 og POU3F4 gena voru magnaðar upp með fjölliðunarhvarfi og leitað var að erfðabreytileikum í þeim með svokallaðri EMD tækni. Niturbasaraðir þeirra útraða, sem sýndu merki um erfðabreytileika, voru síðan ákvarðaðar.
Niðurstöður og ályktun: Með ofangreindum aðferðum greindust fjórir erfðabreytileikar í geni Cx26. Víkjandi samsætan Cx26 35delG fannst hjá einum arfhreinum og öðrum arfblendnum einstaklingi. Hjá arfblendna 35delG einstaklingnum fannst einnig þriggja basa brottfall 358-360delGAG (E), en sú samsæta er einnig víkjandi. Hjá einum arfblendum einstaklingi fundust niturbasaskipti, T verður C í stöðu 101 (M34T) og er talið að sá erfðabreytileiki valdi ríkjandi heyrnarskerðingu með breytilegri sýnd. Þá greindist áður óþekktur erfðabreytileiki í 5'-enda Cx26 gensins hjá einum einstaklingi með fulla heyrn, T verður G í stöðu -63, og er klínískt vægi hans óvisst. Engir erfðabreytileikar greindust í POU3F4 geni.
English Summary |
| Ólafsson Í, Hjaltadóttir G, Cook E, Þórisson HM, Eiríksdóttir G, Petersen H Hereditary hearing impairment. Mutation analysis of connexin 26 and POU3F4 genes in Icelanders with nonsyndromic hearing impairment Læknablaðið 2000; 86: 833-9 Aims: Mutations in the connexin 26 (Cx26) gene have recently been shown to be a major cause of hereditary nonsyndromic sensorineural hearing impairment in Caucasians. Studies indicate that approximately 10-30% of all childhood deafness are due to Cx26 mutations and the most frequently observed mutation is Cx26 35delG. Mutations in the POU3F4 are the most common cause of X-linked nonsyndromic hereditary hearing impairment. The aim of our study was to determine presence and type of Cx26 and POU3F4 mutations in an Icelandic cohort with nonsyndromic hearing impairment. Material and methods: All 15 individuals participating in the study, fulfilled the criteria of severe congenital nonsyndromic hearing impairment of unknown cause and the hearing loss was documented by audiologic testing in a clinical facility. Eleven had a family history and four were sporadic cases. All exons of the Cx26 and POU3F4 genes were amplified using PCR and six pairs of primers. The amplified DNA fragments were screened for sequence variations using enzymatic mutation detection and the nucleotide sequence of fragments showing signs of variation was determined. Results and conclusions: Using the methods described above four distinct sequence variations were detected in the Cx26 gene. The 35delG allele causing hereditary recessive hearing impairment was identified in one homozygous and one heterozygous individual. The heterozygous 35delG individual was also shown to carry the recessive allele 358-360delGAG (E). A missense mutation, 101Tð C (M34T), supposed to cause autosomal dominant form of hearing impairment with variable penetrance, was detected in one heterozygous individual. A novel sequence variation without known clinical significance, -63Tð G, was found in the 5'-noncoding sequence in one control sample. No mutations were detected in the POU3F4 gene. Key words: hereditary hearing impairment, connexin 26, POU3F4. Correspondence: Ísleifur Ólafsson. E-mail: isleifur@shr.is |
Inngangur
Erlendar rannsóknir hafa sýnt að um það bil eitt barn af hverjum 1000 hafa mikla meðfædda heyrnarskerðingu, en mun fleiri fæðast með vægari form (1-3). Algengi heyrnarskerðingar eykst síðan eftir miðjan aldur, en um 4% 45 ára manna hafa verulega skerta heyrn og við 70 ára aldur er hlutfallið orðið um 30% (1).Orsök heyrnarskerðingar hjá nýfæddum er talin vera af arfgengum toga í um 60-70% tilvika (4). Um 30% af arfgengri heyrnarskerðingu eru tengd ýmsum heilkennum (syndromes), en 70% eru án nokkurra annarra klínískra einkenna (5-7). Talið er að arfgeng heyrnarskerðing án tengsla við heilkenni erfist í 75% tilfella víkjandi, um 20-25% ríkjandi og í 1-5% tilvika er um kynbundnar eða hvatberaerfðir að ræða (8,9). Hvað varðar heyrnarskerðingu, sem kemur á efri árum og er oft tengd umhverfisáhrifum, svo sem hávaða eða lyfjagjöf, þykir sýnt að arfgengir þættir koma þar einnig við sögu (10-12).
Þekking manna á arfgengum þáttum meðfæddrar heyrnarskerðingar án tengsla við heilkenni hefur aukist hratt á síðustu árum (5,6,9). Meingenaleit hjá fjölskyldum frá öllum heimsins hornum hafa sýnt að erfðabreytingar á tæplega 70 mismunandi litningasetum geta komið við sögu, þegar um er að ræða heyrnarskerðingu án tengsla við heilkenni (13). Um 30 þeirra eru víkjandi set, önnur 30 eru ríkjandi, átta eru á X litningi og tvö eru í erfðaefni hvatbera. Sextán þessara gena hafa nú verið einangruð og raðgreind og er hlutverk þeirra prótína sem genin skrá mjög mismunandi (6,13). Þau geta verið hluti jónarásar, utanfrumuprótín, frumugrindarprótín og afritunarþættir, en nokkur þeirra hafa óþekkt hlutverk.
Orsök arfgengrar heyrnarskerðingar án tengsla við heilkenni er langoftast að finna í geni konnexíns 26 (Cx26), sem er staðsett á litningi 13q11 (14-17). Talið er að erfðabreytileikar í þessu geni valdi 20-70% tilfella af meðfæddri heyrnarskerðingu (18-23). Einn erfðabreytileiki er áberandi algengasta orsök arfgengrar heyrnarskerðingar meðal Evrópubúa, en það er Cx26 35delG (brottfall á niturbasanum gúanín í stöðu 35 í geni Cx26) (18,19,24). Hjá ítölskum og spönskum einstaklingum með heyrnarskerðingu af völdum erfðabreytileika í Cx26 geni er þennan eina erfðabreytileika að finna hjá um 85% (19). Hingað til hafa um það bil 35 erfðabreytileikar sem valda heyrnarskerðingu fundist í Cx26 geni og er algengi þeirra mjög mismunandi eftir þýðum (25).
Prótínið, sem Cx26 genið skráir, er ýmist kallað konnexín 26 eða GJB2 (gap junction protein b2). Það tilheyrir stórri fjölskyldu þróunarlega skyldra himnuprótína og eru að minnsta kosti 16 mismunandi gerðir konnexína að finna í manninum (26,27). Þau hafa þó öll svipaða byggingu og hlutverk. Sex prótínsameindir konnexína tengjast sín á milli í tvílagi frumuhimna og mynda sívalningslaga rás í gegnum þær. Slíkar rásir eru kallaðar konnexón. Konnexón geta ýmist verið samsettar úr einni gerð konnexína (homeomeric) eða úr tveimur eða fleiri mismunandi gerðum (heteromeric). Konnexón tveggja aðliggjandi frumna tengjast síðan sín á milli og mynda smárásir (gap junction channels) milli frumna. Jónir og minni sameindir geta borist í gegnum þessar rásir og koma þær þannig á mjög nánum samskiptum frumnanna. Smárásirnar eru ýmist opnar eða lokaðar og stjórnast það af virkni í efnaskiptum viðkomandi frumna (26,27). Nýlega hefur verið sýnt fram á að erfðabreytileikar í genum konnexína 30 og 31 geta einnig á svipaðan hátt og í Cx26 orsakað heyrnarskerðingu án tengsla við heilkenni (28,29).
POU3F4 genið er staðsett á X-litningi, nánar tiltekið Xq21 (30-32). Erfðabreytileikar í geninu eru algengasta þekkta orsök heyrnarskerðingar sem erfist kynbundið (32-34). Tölvusneiðmyndir af innra eyra einstaklinga með heyrnarskerðingu af völdum erfðabreytileika í POU3F4 hafa í sumum tilfellum sýnt óeðlilega útvíkkun á innri heyrnargangi og við skurðaðgerðir hefur komið í ljós hækkaður vökvaþrýstingur í utanvessa (perilympha) og að ístað miðeyrans er gróið fast í sporgati (oval window) (30,35). Prótínið POU3F4 er afritunarþáttur, sem tilheyrir stórri fjölskyldu þróunarlega skyldra afritunarþátta, sem nefnist POU, og er POU3F4 genið tjáð í mörgum frumutegundum, þó einkum í taugavef (32).
Markmið þessarar rannsóknar var að kanna hvort og hvaða erfðabreytileikar í genum konnexíns 26 og í POU3F4 gætu verið orsök heyrnarskerðingar hjá Íslendingum með meðfædda heyrnarskerðingu og án tengsla við heilkenni.
Efniviður og aðferðir
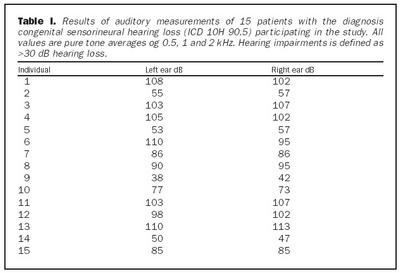
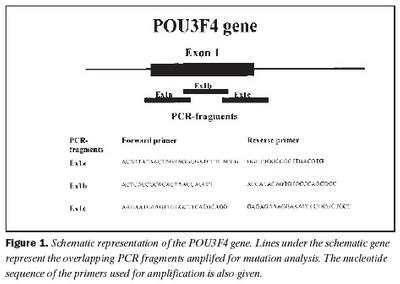
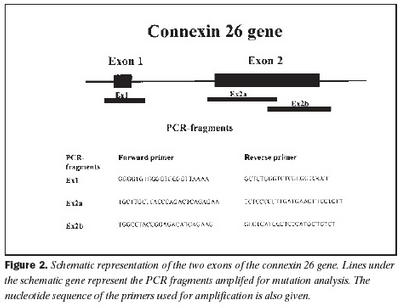
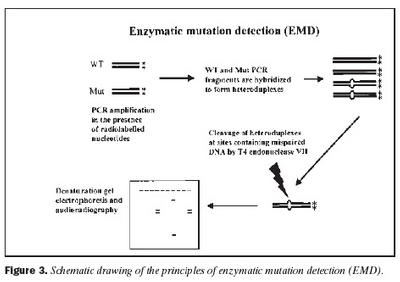
Úrtak: Í samvinnu við Félag heyrnarlausra, Foreldrafélag heyrnarlausra og Heyrnarhjálp var leitað eftir þátttöku með því að senda út kynningarbréf. Allir þátttakendur undirrituðu eyðublað um upplýst samþykki. Alls fengust 15 þátttakendur, sem uppfylltu þau skilyrði að hafa meðfædda skyntaugaheyrnarskerðingu (sensorineural hearing impairment) af óþekktum toga (ICD 10 H 90.5) og að heyrnarskerðingin væri staðfest með heyrnarmælingu hjá Heyrnar- og talmeinastöð Íslands eða á háls-, nef og eyrnadeild Landspítala Fossvogi. Meðalaldur þátttakenda var 29 ára (8-69 ára) og voru sjö karlar og átta konur. Ellefu þátttakendur höfðu ættarsögu um heyrnarleysi, en fjórir voru stök tilfelli. Tafla I sýnir niðurstöður heyrnarmælinga á báðum eyrum hjá þátttakendum. Rannsóknin var samþykkt af Tölvunefnd Dómsmálaráðuneytis og Vísindasiðanefnd Sjúkrahúss Reykjavíkur. Aðferðir: Erfðaefni þátttakenda var eingangrað úr bláðæðablóði eins og áður hefur verið lýst (36). Niturbasaraðir gena Cx26 og POU3F4 voru fengnar af heimasíðu National Center for Biotechnology Information í Bandaríkjunum (http://www. ncbi.nlm.nih.gov) (32,37,38). Fákirni voru valin með hjálp GenetoolTM 1.0 forritsins (Biotools Incorporated, Edmonton, Kanada) og þau pöntuð frá TAG Copenhagen A/S í Kaupmannahöfn. Genabygging Cx26 og POU3F4, niturbasaröð fákirna, sem notuð voru við fjölliðunarhvörf (polymerase chain reaction, PCR) og staðsetning þeirra í genunum eru sýndar á myndum 1 og 2. Fjölliðunarhvörf á einstökum DNA-bútum genanna voru gerð á hefðbundinn hátt nema hvað geislavirkt a-33P dATP var haft með í hvarfefnablöndu til að merkja fjölfölduðu bútana. Skimað var eftir erfðabreytileikum í DNA-bútum með svokallaðri EMD-aðferð (enzymatic mutation detection) og var hvarfefnasettið PassportTM frá Amersham-Pharmacia Biotech, Kaupmannahöfn notað (39). Einfölduð mynd til skýringar á aðferðafræði EMD er sýnd á mynd 3. Eftir að misþátta (heteroduplex) DNA-bútar höfðu verið meltir með T4 endónúkleasa VII, voru þeir rafdregnir í 6% raðgreiningargeli. Að loknum rafdrætti var röntgenfilma lögð að gelinu og hún framkölluð um sólarhring seinna. Niturbasaröð þeirra DNA-búta, sem sýndu merki um að hafa verið klofnir af T4 endónúkleasa VII vegna mispörunar, var ákvörðuð með því að nota sömu fákirni og við fjölföldunina,
a-33P dATP, a-33P dCTP, hvarfefnasettið ThermoSequenaseTM frá Amersham Pharmacia Biotech, Kaupmannahöfn og 6% DNA raðgreiningargel.
Niðurstöður
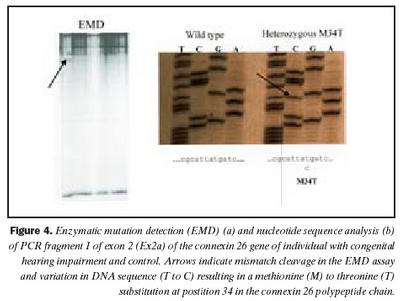
Cx26 gen: Við leit að erfðabreytileikum með EMD aðferð komu fram merki um erfðabreytileika í fjölfölduðum DNA bútum Cx26 gens hjá fjórum einstaklingum. Dæmi um greinilega jákvæða EMD skimun fyrir erfðabreytileika má sjá í mynd 4. Í bút Ex2a í Cx26 geni komu fram merki um þrjá mismunandi breytileika. Sams komar EMD mynstur í rafdráttargeli fundust hjá þátttakendum númer átta og níu (tafla I), annað mynstur fannst hjá þátttakenda númer tvö og það þriðja hjá einstaklingi sem notaður var sem viðmiðunarsýni. Í bút Ex2b fannst merki um erfðabreytileika hjá þátttakenda númer níu. Raðgreining á DNA bút Ex2a hjá einstaklingi númer tvö sýndi arfblendni fyrir erfðabreytileika, þar sem niturbasinn C kemur í stað T í stöðu 101 í geninu (mynd 4). Þessi breytileiki hefur í för með sér amínósýruskipti í fjölpeptíðkeðju Cx26, þar sem amínósýran treónín kemur í stað meþíóníns í stöðu 34 (M34T). Erfðabreytileikanum M34T hefur áður verið lýst en hann er talinn valda ríkjandi erfðamynstri, þó með mjög breytilegri sýnd (17,21). Niðurstöður heyrnarmælingar þessa einstaklings sýndu verulega skerta heyrn (um 55dB), en hann hafði ekki fjölskyldusögu um heyrnarskerðingu.
Raðgreining á PCR bút Ex2a hjá einstaklingum númer átta og níu sýndi brottfall á G í stöðu 35 í geni Cx26 (Cx26 del35G), en þessi erfðabreytileiki veldur arfgengri, víkjandi heyrnarskerðingu og er algengasta orsök afrgengrar heyrnarskerðingar eins og áður sagði (mynd 5) (19,20,24). Cx26 del35G veldur lesrammahliðrun (frameshift) og síðan stöðvun á þýðingu mRNA. Þessi erfðabreytileiki veldur heyrnarskerðingu með víkjandi erfðamáta. Einstaklingur númer átta var arfhreinn, en númer níu arfblendinn (mynd 5). Því mátti búast við að finna annan erfðabreytileika í geni Cx26 hjá einstaklingi númer níu og gaf EMD merki um erfðabreytileika í bút Ex2b. Niðurstöður heyrnarmælingar hjá einstaklingi númer átta sýndu heyrnarskerðingu um 90 dB og var saga um skerta heyrn hjá nánum ættingjum.
Raðgreining á PCR bút Ex2b hjá einstaklingi númer níu sýndi brottfall á þremur niturbösum í stöðu 358-360 í geni Cx26 (358-360delGAG) (mynd 6). Erfðabreytileikinn veldur brottfalli á heilum lesramma í mRNA og því einnig brottfalli á einni amínósýru í fjölpeptíðkeðju Cx26 prótínsins, en það er glútamiksýra í stöðu 119 (E). Þessum erfðabreytileika hefur áður verið lýst sem orsök arfgengrar heyrnarskerðingar með víkjandi erfðamáta (20,40). Einstaklingur númer átta, sem ber erfðabreytileikann 358-360delGAG á arfblendnu formi, ber einnig Cx 35delG og hefur því svokallaða samsetta arfblendni (combined heterozygosity). Niðurstöður heyrnarmælingar einstaklings númer níu sýndu heyrn um 40dB og hafði hann fjölskyldusögu um heyrnarskerðingu.
Raðgreining á DNA bút Ex2a úr Cx26 geni, sem gaf merki um erfðabreytileika, en var úr einstaklingi sem notaður var sem viðmiðunarsýni, leiddi í ljós arfblendni fyrir niturbasaskiptum þar sem T verður C í stöðu -63 (-63Tð C) (mynd 7). Þar sem þessi niturbasi er utan við kóðandi svæði gensins hefur erfðabreytileikinn ekki í för með sér amínosýruskipti í fjölpeptíðkeðju eða truflun í lesramma. Þessum erfðabreytileika hefur ekki verið lýst áður, en þar sem arfberinn hefur fulla heyrn er óvíst hvort erfðabreytileikinn hafi klíníska þýðingu.
POU3F4 gen: Þrátt fyrir ítrekaða skimun með EMD aðferð fundust ekki merki um erfðabreytileika í POU3F4 geni hjá þátttakendum.
Umræða
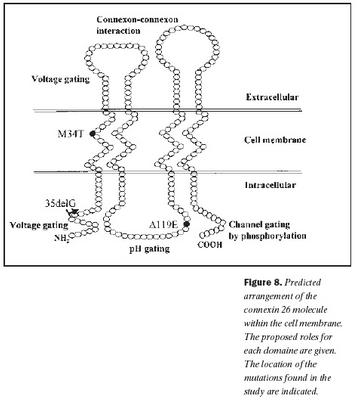
Niðurstöður þessarar rannsóknar sýna að, að minnsta kosti þrír erfðabreytileikar í geni Cx26 valda heyrnarskerðingu hér á landi, 35delG, 101Tð C, og 358-360delGAG. Einfölduð mynd af byggingu Cx26 og staðsetning þeirra erfðabreytileika, sem fundust í rannsókninni, er sýndar á mynd 8. Auk þess fannst nýr erfðabreytileiki í Cx26 geninu hjá viðmiðunareinstaklingi, -63Tð C, sem er staðsettur utan við kóðandi svæði þess gens og er óvíst með klínískt vægi hans. Engir erfðabreytileikar fundust í POU3F4 geni.Erfðabreytileikar í Cx26 geni er algengasta þekkta orsök meðfæddrar, arfgengrar heyrnarskerðingar án tengsla við heilkenni (18,19,24). Þessi erfðabreytileiki veldur lesrammahliðrun í þýðingu og því myndast ekki fjölpeptíðkeðja Cx26 (mynd 8). Klínísk einkenni arfhreinna einstaklinga með 35delG geta verið frá tiltölulega vægri heyrnarskerðingu upp í mikla (40-42). Við rannsókn á 82 fjölskyldum frá Ítalíu og Spáni með víkjandi form arfgengrar heyrnarskerðingar og 54 heyrnarskertum einstaklingum án fjölskyldusögu, reyndust 49% fjölskyldnanna og 37% stakra einstaklinga bera erfðabreytileika í geni Cx26 og var um að ræða 35delG í 85% tilvika (19). Í sömu rannsókn var arfberatíðni Cx26 35delG könnuð í 280 manna úrtaki með eðlilega heyrn og var hún 3,2%. Fjölþjóðleg evrópsk rannsókn sýndi að arfberatíðni í Suður-Evrópu var á bilinu 2,2-3,4% og í Mið-Evrópu 0,5-2,2% (24). Mikill munur var á arfbertíðni 35delG í þremur ríkjum í Norður-Evrópu, en þar hafði Eistland 4,4% Danmörk 2,1% og Noregur 0,5%. Í bandarískri rannsókn á arfberatíðni reyndist hún 2,5% fyrir Cx26 35delG, en 3,0% fyrir alla erfðabreytileika í geni Cx26, sem valda víkjandi arfgengri heyrnarskerðingu (43). Meðal Gyðinga er tíðni 35delG mun lægri eða um 0,7%, en tíðni annars erfðabreytileika, Cx26 167delT, er mun hærri eða um 4% (44,45). Hjá Afríkubúum er tíðni 35delG lág, en sýnt hefur verið að tíðni einstakra erfðabreytileika, svo sem Cx26 R143W, getur verið mjög há svæðisbundið (46,47). Hjá Japönum er Cx26 235delC erfðabreytileikinn algengastur en 35delG hefur ekki fundist (48).
Ekki er hægt að draga ályktanir um algengi einstakra erfðabreytileika í Cx26 geni frá rannsókn okkar, þar sem úrtak er of lítið. Niðurstöðurnar benda þó til að tíðni Cx26 35delG erfðabreytileikans sé ekki eins algeng og meðal þjóða við Miðjarðarhafið.
Erfðabreytileikinn 101Tð C eða M34T var fyrsti erfðabreytileikinn í Cx26 geni sem fannst og er talinn valda ríkjandi erfðamáta með mjög breytilegri sýnd (17,21). Þannig geta arfblendnir einstaklingar ýmist verið með eðlilega heyrn eða verulega skerta. Erfðabreytileikinn 101Tð C veldur amínósýruskiptum, þar sem treónín kemur í stað meþíóníns í stöðu 34 í fjölpeptíðkeðju Cx26. Þessi amínósýra er staðsett í þeim hluta fjölpeptíðskeðjunnar sem gengur í gegnum frumuhimnuna og víxlverkar við önnur konnexín (mynd 8). Með tjáningu villigerðar og M34T Cx26 í ræktuðum frumum hefur verið sýnt fram á að amínósýruskipin trufla þessa víxlverkun þannig að konnexín geta ekki tengst sín á milli og myndað konnexón og rás gegnum frumuhimnur (49,50). Rannsóknir á arfberatíðni erfðabreytileikans 101Tð C, hafa sýnt að hann er nokkuð algengur meðal Evrópubúa eða á bilinu 0,5-1,5% (21,51).
Erfðabreytileikanum Cx26 358-360delGAG var fyrst lýst í tveimur áströlskum systrum með samsetta arfblendni með R148P og síðan nýlega í einum Ítala með samsetta arfblendni með 167delT (20,40). Þessi erfðabreytileiki veldur víkjandi erfðamáta. Sá einstaklingur, sem fannst í þessari rannsókn, var með samsetta arfblendni með 35delG og tiltölulega væga heyrnarskerðingu. Erfðabreytileikinn veldur brottfalli á einni súrri amínósýru (glútamíni í stöðu 118), sem er staðsett í þeim hluta fjölpeptíðkeðju Cx26, sem er innan frumuhimnunnar og hefur með pH-háða stjórnun á smárásinni að gera (mynd 8) (26,27).
Erfðabreytileikanum -63Tð C, sem fannst hjá einum viðmiðunareinstaklingi með fulla heyrn, hefur ekki verið lýst áður. Hann er staðsettur utan við kóðandi svæði gensins og veldur því ekki amínósýruskiptum. Hann er að öllum líkindum án klínískrar þýðingar, en gæti þó hugsanlega haft áhrif á hversu hratt mRNA Cx26 er brotið niður.
Í rannsókninni fundust engir erfðabreytileikar í geni POU3F4, en erfðabreytileikar í þessu geni eru algengasta orsök heyrnarskerðingar, sem erfist kynbundið.
Með vaxandi þekkingu á arfgengum þáttum heyrnarskerðingar og þróun aðferða til að greina þá, opnast möguleikar á að greina heyrnarskerðingu hjá börnum fyrr og leggja mat á horfur. Miklivægt er að greina snemma alvarlega heyrnarskerðingu hjá börnum, þar sem fyrsta ár ævinnar er afgerandi í málþroska þeirra. Þar sem margir erfðabreytileikar í mörgum mismunandi genum geta orsakað heyrnarskerðingu þarf að þekkja þá erfðabreytileika sem eru algengastir í viðkomandi þýðum. Þessi rannsókn sýndi að, að minnsta kosti þrír mismunandi erfðabreytileikar í Cx26 geni valda heyrnarskerðingu á Íslandi. Hún sýndi einnig að þar sem margir þátttakenda höfðu fjölskyldusögu er full ástæða til þess að leita skýringa í fleiri genum.
Þegar tekin er ákvörðun um að nota erfðafræðilegar aðferðir við greiningu á orsök heyrnarskerðingar þarf hafa í huga eftirtalin þrjú atriði.
Í fyrsta lagi eru gen sem tengjast heyrnarskerðingu mjög mörg og niturbasaröð flestra þeirra hefur enn ekki verið ákvörðuð. Þetta gerir það að verkum að ekki er hægt að bjóða upp á erfðagreiningu, sem gefur öruggt svar við því hvort um sé að ræða arfgenga þætti eða ekki.
Í öðru lagi getur það verið tæknilega erfitt og afar kostnaðarsamt að finna þann erfðabreytileika sem veldur heyrnarskerðingunni þar sem leita þarf í tugum gena. Með aukinni þekkingu á erfðafræði heyrnarskerðingar og tæknilegum framförum má þó búast við að í framtíðinni verði erfðagreining öruggari, auðveldari og ódýrari en hún er nú.
Í þriðja lagi skal hafa í huga að verulegur hluti heyrnarskertra er ekki samþykkur því að erfðagreiningu sé beitt við heyrnarskerðingu.
Í ljósi ofangreindra atriða og þess, að meðferð, sem beinist sértækt að orsökum arfgengrar heyrnarskerðingar, hefur ekki komið fram, ber lækni að meta ábendingu fyrir erfðagreiningu í hverju einstöku tilfelli fyrir sig og nýta sér erfðaráðgjöf faglærðra eftir kostum.
Þakkir
Höfundar vilja þakka öllum þátttakendum í rannsókninni, Félagi heyrnarlausra, Foreldrafélagi heyrnarlausra, Heyrnarhjálp, Málfríði Gunnarsdóttur, Einari Sindrasyni yfirlækni og Heyrnar- og talmeinastöð Íslands. Verkefnið var styrkt af Vísindasjóði Sjúkrahúss Reykjavíkur.
Heimildir
1. Nadol JB. Hearing loss. N Engl J Med 1993: 15; 1092-102.2. Van Narden K, Decoufle P, Caldwell K. Prevalence and characteristics of children with serious hearing impairment in metropolitan Atlanta, 1991-1993. Pediatrics 1999; 103: 570-5.
3. Morton NE. Genetic epidemiology of hearing impairment. Ann NY Acad Sci 1991; 46: 95-105.
4. Marazita ML, Ploughman LM, Rawlings B, Remington E, Arnos KS, Nance WE. Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am J Med Genet 1993; 46: 486-91.
5. Petit C. Genes responsible for human hereditary deafness: symphony of a thousand. Nature Genet 1996; 14: 385-91.
6. Skvorak Giersch AB, Morton CC. Genetic causes of nonsyndromic hearing loss. Curr Op Ped 1999; 11: 551-7.
7. Van Camp G, Willems PJ, Smith RJH. Non-syndromic hearing impairment: unparalleled heterogeneity. Am J Hum Genet 1997; 60: 758-64.
8. Tomaski SM, Grundfast KM. A stepwise approach to the diagnosis and treatment of hereditary hearing loss. Pediatr Clin North Am 1999; 46: 35-49.
9. Grundfast KM, Atwood JL, Choung D. Genetics and molecular biology of deafness. Otolaryngol Clin North Am 1999; 32: 1067-88.
10. Sill AM, Stick MJ, Prenger VL, Phillips SL, Boughman JA, Arnos KS. Genetic epidemiologic study of hearing loss in an adult population. Am J Med Genet 1994; 54: 149-53.
11. O'Neill ME, Marinetta J, Nihimura D, Wayne S, Van Camp G, Van Laer L, et al. A gene for autosomal dominant late-onset progressive non-syndromic hearing loss, DFNA10, maps to chromosome 6. Hum Mol Genet 1996; 5: 853-6.
12. Morell RJ, Friderici KH, Wei S, Elfenben JL, Friedman TB, Fisher RA. A new locus for late-onset, progressive hereditary hearing loss DFNA20 maps to 17q25. Genomics 2000; 63: 1-6.
13. Van Camp G, Smith RJH. Hereditary Hearing Loss Homepage. World Wide Web URL: http://dnalab-www.uia.ac.be/dnalab/hhh/
14. Guilford P, Ben Arab S, Blanchard S, Levilliers J, Weissenbach J, Belkahia A, et al. A non-syndromic form of neurosensory, recessive deafness maps to the periocentromeric region of chromosome 13q. Nature Genet 1994; 6: 24-8.
15. Brown KA, Janjua AH, Karbani G, Parry G, Noble A, Crockford G, et al. Linkage studies of non-syndromic recessive deafness (NSRD) in a family originating from the Mipur region of Pakistan maps DFNB1 centromeric to D13S175. Hum Mol Genet 1996; 5: 169-75.
16. Gasparini P, Estivill X, Volpini V, Totaro A, Castellvi-Bel S, Govea N, et al. Linkage of DFNB1 to non-syndromic neurosensory autosomal recessive deafness in Mediterranean families. Eur J Hum Gent 1997; 5: 83-8.
17. Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997; 387: 80-3.
18. Zelante L, Gasparini P, Estivill X, Melchionda S, D'Agruma L, Govea N, et al. Connexin 26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum Mol Genet 1997; 6: 1605-9.
19. Estevill X, Fortina P, Surray S, Rabionet R, Melchionda S, D'Agruma L, et al. Connexin-26 mutations in sporadic and inherited sensoryneural deafness. Lancet 1998; 351: 394-8.
20. Denoyelle F, Weil D, Maw MA, Wilcox SA, Lench NJ, Allen-Powell DR, et al. Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Hum Mol Genet 1997; 6: 2173-7.
21. Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, et al. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet 1998; 62: 792-9.
22. Scott DA, Kraft ML, Carmi R, Ramesh A, Elbedour K, Yairi Y, et al. Identification of mutations in the connexin 26 gene that cause autosomal recessive nonsyndromic hearing loss. Hum Mut 1998; 11: 387-94.
23. Lench N, Housman M, Newton V, Van Camp G, Mueller R. Connexin-26 mutations in sporadic non-syndromal sensorineural deafness. Lancet 1998; 351: 415.
24. Gasparini P, Rabionet R, Barbujani G, Melchionda S, Petersen M, Brondum-Nilsen K, et al. High carrier frequency of the 35delG deafness mutation in European populations. Genetic analysis consortium of GJB2 35delG. Eur J Hum Genet 2000; 8: 19-23.
25. The Human Gene Mutation Database, Cardiff: http://www.uwcm.ac.uk/uwcm/mg/ns
26. Kumar NM, Gilula NB. The gap junction communication channel. Cell 1996; 84: 381-8.
27. Yeager M, Unger VM, Falk MM. Synthesis assembly and structure of gap junction intercellular channels. Curr Opin Struct Biol 1998; 8: 517-24.
28. Grifa A, Wagner CA, D'Ambrosio L, Melchionda S, Bernardi F, Lopez-Bigas N, et al. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat Genet 1999; 23: 16-8.
29. Xia J, Liu C, Tang B, Pan Q, Huang L, Dai HP, et al. Mutations in the gene encoding gap junction protein b-3 associated with autosomal dominant hearing impairment. Nature Genet 1998; 20: 370-3.
30. Brunner HG, van Bennekom A, Lambermon EM, Oei TL, Cremers WR, Wieringa B, et al. The gene for X-linked progressive mixed deafness with perilymphatic gusher during stapes surgery (DFN3) is linked to PGK. Hum Genet 1988; 80: 337-40.
31. Reardon W, Middleton-Price HR, Sandkuijl L, Phelps P, Bellmann S, Luxon L, et al. A multipedigree linkage study of X-linked deafness: linkage to Xq13-q21 and evidence for genetic heterogeneity. Genomics 1991; 11: 885-94.
32. de Kok YJM, Van der Maarel SM, Bitner-Glindzicz M, Huber I, Monaco AP, Malcolm S, et al. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science 1995; 267: 685-8.
33. de Kok YJ, Cremers CW, Ropers HH Cremers FP. The molecular basis of X-linked deafness type 3 (DFN3) in two sporadic cases: identification of a somatic mosaicism for a POUF4 missense mutation. Hum Mutat 1997; 10: 207-11.
34. Bitner-Glindzicz M, Turnpenny P, Höglund P, Kääriäinnen H, Sankila E-M, van der Maarel SM, et al. Further mutations in Brain 4 (POU3F4) clarify the phenotype in the X-linked deafness, DFN3. Hum Mol Genet 1995; 4: 1467-9.
35. Phelps PD, Reardon W, Pembrey ME, Bellman S, Luxom L. X-linked deafness, stapes gushers and a distinctive defect of the inner ear. Neuroradiolgy 1991; 33: 326-30.
36. Bell GI, Karam JH, Rutter W. Polymorphic DNA region adjacent to the 5'-end of the human insulin gene. Proc Natl Acad Sci USA 1981; 78: 5759-63.
37. Lee SW, Tomasetto C, Paul D, Keyomarsi K, Sanger R. Transcriptional downregulation of gap-junction proteins block junctional communication in human mammary tumor cell lines. J Cell Biol 1992; 118: 1213-21.
38. Kiang DT, Jin N, Tu ZJ, Lin HH. Upstream genomic sequence of the human connexin 26 gene. Gene 1997; 199: 165-71.
39. Del Tito BJ, Poff III HE, Novotny MA, Cartledge DM, Walker RI 2nd, Earl CD, et al. Automated fluorescent analysis procedure for enzymatic mutation detection. Clin Chem 1998; 44: 731-9.
40. Murigia A, Orzan E, Polli R, Martella M, Vinanzi C, Leonardi E, et al. Cx26 deafness: mutation analysis and clinical variability. J Med Genet 1999; 36: 829-32.
41. Denoyelle F, Marlin S, Weil D, Moatti L, Chauvin P, Garabedian EN, et al. Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin 26 gene defect: implications for genetic counselling. Lancet 1999; 353: 1298-303.
42. Cohn ES, Kelley PM, Fowler TW, Gorga MP, Lefkowitz DM, Kuehn HJ, et al. Clinical studies of families with hearing loss attributable to mutations in the connexin 26 gene. Pediatrics 1999; 103: 546-50.
43. Green GE, Scott DA, McDonald JM, Woodworth GG, Sheffield VC, Smith RJ. Carrier rates in the the midwestern United States of GJB2 mutation causing inherited deafness. JAMA 1999; 281: 2211-6.
44. Morell RJ, Kim HJ, Hood LJ, Goforth L, Friderici K, Fisher R, et al. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic decessive deafness. N Engl J Med 1998; 339: 1500-5.
45. Sobe T, Erlich P, Berry A, Korostichevsky M, Vreugde S, Avraham KB, et al. High frequency of the deafness-associated 167delT mutation in the connexin 26 (GJB2) gene in Israeli Ashkenazim. Am J Med Genet 1999; 86: 499-500.
46. Brobby GW, Muller-Myhsok B, Horstmann RD. Connexin 26 R143W mutation associated with recessive nonsyndromic sensorineural deafness in Africa. N Engl J Med 1998; 338: 548-50.
47. Brooker JK, Zhou Z, Silvermen LM, Rohlfs EM. Frequency of GJB2 (connexin 26) in African American and Caucasian populations in North Carolina. Am J Hum Genet 1999; 65/Suppl: A198.
48. Kudo T, Ikeda K, Kure S, Matsubara Y, Oshima T, Watanabe Ki, et al. Novel mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population. Am J Hum Genet 2000; 90: 141-5.
49. Martin PE, L Coleman S, Casalotti SO, Forge A, Evans WH. Properties of connexin 26 gap junctional proteins derived from mutations associated with non-syndromal heriditary deafness. Hum Mol Genet 1999; 8: 2369-76.
50. White TW, Deans MR, Kelsell DP, Paul DL. Connexin mutations in deafness. Nature 1998; 394: 630-1.
51. Scott DA, Kraft ML, Stone EM, Sheffield VC, Smith RJH. Connexin mutations and hearing loss. Nature 1998; 391: 32.