
Fræðigreinar
Arfbundin kólesterólhækkun. Yfirlit yfir stöðu þekkingar og árangur markvissrar leitar á Íslandi.
Ágrip
Arfbundin hækkun á kólesteróli (familial hypercholesterolemia, FH) er erfðasjúkdómur sem orsakar ævilanga hækkun á kólesteróli og hefur í för með sér mjög aukna hættu á kransæðasjúkdómi. Lyfjameðferð með kólesteróllækkandi lyfjum er oftast árangursrík og benda rannsóknir til að meðferðin dragi verulega úr hættu á kransæðasjúkdómi. Sjúkdómsgreiningin byggir á kólesterólmælingu einstaklingsins og nánustu ættingja eða á erfðaprófi. Algengi sjúkdómsins er áætlað 1/500.
Hér á landi hefur greinst tæpur helmingur þeirra sem áætlað er að hafi arfbundna kólesterólhækkun (rúmlega 200 manns). Þrjár stökkbreytingar hafa fundist sem valda sjúkdómnum í Íslendingum og er ein langalgengust og áætlað að hún valdi 60% af arfbundinni hækkun á kólesteróli hér á landi.
Í þessari grein er gefið stutt yfirlit yfir stöðu þekkingar á arfbundinni kólesterólhækkun, einkum með tilliti til hættu á kransæðasjúkdómi og gerð grein fyrir þeim árangri sem náðst hefur í leit að sjúkdómnum hér á landi. Einnig er kynnt átak Hjartavendar til að finna ógreinda einstaklinga með sjúkdóminn.
English Summary |
| Þórsson B, Guðnason V, Þorvaldsdóttir G, Sigurðsson G Familial hypercholesterolemia in Iceland. Review and outcome of family screening Læknablaðið 2001; 87: 513-8 Familial hypercholesterolemia is a genetic disorder causing lifelong elevation of cholesterol and severely increased risk of coronary heart disease. Cholesterol lowering drug treatment is usually effective and clinical trials indicate a substantial lowering of risk, thanks to treatment. The diagnosis is based on cholesterol level in the individual and in his/her closest relatives or on a genetic test. The prevalence is estimated 1/500. In Iceland nearly half of the estimated number of individuals with familial hypercholesterolemia has been diagnosed (about 200). Three mutations have been identified that cause familial hypercholesterolemia in the Icelandic population and the most common one is estimated to cause 60% of the disease in Iceland. In this paper we give a short review of the literature on familial hypercholesterolemia especially regarding risk of coronary heart disease and describe the results of screening for the disease in Iceland. Also we present a new campaign to find undiagnosed individuals with the disease. Key words: familial hypercholesterolemia, coronary heart disease, screening. Correspondence: Bolli Þórsson. E-mail: bolli@hjarta.is |
Inngangur
Undanfarna tvo áratugi hefur verið leitað með kerfisbundnum hætti að einstaklingum með arfbundna kólesterólhækkun (familial hypercholesterolemia, FH) á göngudeild háþrýstings og blóðfitu Landspítala Hringbraut. Áætlað er að tæpur helmingur einstaklinga með arfbundna kólesterólhækkun á Íslandi hafi þegar greinst á deildinni. Vegna margfalt aukinnar hættu á kransæðasjúkdómi hjá einstaklingum með arfbundna kólesterólhækkun hefur Alþjóðaheilbrigðisstofnunin hvatt til átaks í greiningu og meðhöndlun sjúkdómsins (1). Hjartavernd hefur nú hafið slíkt átak þar sem leitast er við að finna sem flesta einstaklinga með þennan sjúkdóm í þeim tilgangi að bjóða þeim viðeigandi meðferð en einnig verður haldið áfram með faraldsfræðilega og erfðafræðilega rannsókn á sjúkdómnum. Verkefnið er unnið í samvinnu við alþjóðlegu samtökin MEDPED (Make Early Diagnosis Prevent Early Death), sem voru stofnuð til að skrá og aðstoða fólk með arfbundna kólesterólhækkun. Aðilar frá 140 meðferðarstofnunum eru þátttakendur í samtökunum (sjá nánar um samtökin á heimasíðu http://www.medped.org/).Þessari grein er ætlað að gefa stutt yfirlit yfir stöðu þekkingar á arfbundinni kólesterólhækkun einkum með tilliti til hættu á kransæðasjúkdómi og gera grein fyrir árangri leitar að sjúkdómnum hér á landi hingað til.
Almenn atriði varðandi sjúkdóminn
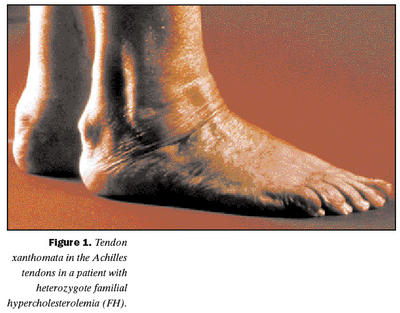
Arfbundin kólesterólhækkun orsakast af stökkbreytingum í geni því sem tjáir viðtaka fyrir kólesterólríku eðlisléttu fituprótíni (low density lipoprotein, LDL) (2). Þessar stökkbreytingar skaða gerð og virkni viðtakans. LDL eru megin flutningsferjur fyrir kólesteról frá lifur til vefja líkamans og er hreinsun þeirra úr plasma skert hjá einstaklingum með arfbundna kólesterólhækkun. Skert virkni LDL viðtakans, sérstaklega á yfirborði lifrarfrumna sem gegna lykilhlutverki í kólesterólefnaskiptunum, orsakar þannig ævilanga hækkun á kólesteróli í blóði. Kólesterólgildi einstaklinga með arfbundna kólesterólhækkun er oftast um tvöfalt hærra en hjá jafnöldrum af sama kyni (3). Langvarandi hátt kólesteról í blóði getur valdið kólesterólútfellingum í ýmsa vefi líkamans og aukið mjög hættu á kransæðasjúkdómi snemma á ævinni og ótímabærum dauðsföllum af þeim sökum. Kólesterólútfellingar í sinar, sérstaklega í hæl- og handarsinar (tendon xanthomata), eru taldar sérkennandi fyrir arfbundna kólesterólhækkun (mynd 1). Sjúkdómurinn erfist ríkjandi og ókynbundið. Af því leiðir að helmingslíkur eru á að afkomandi arfblendins (heterozygote) einstaklings með arfbundna kólesterólhækkun fái sjúkdóminn. Tíðni arfblendinna með sjúkdóminn er álitin vera um einn af hverjum 500 í flestum þýðum en nákvæm tíðni erfðagallans hefur hvergi verið mæld. Á sömu forsendum er algengi arfhreinna með sjúkdóminn áætlað einn af milljón (4).Hætta á kransæðastíflu meðal einstaklinga með arfbundna kólesterólhækkun
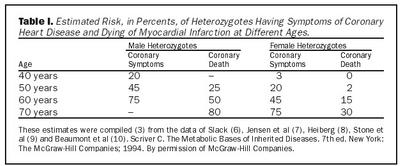
Rannsóknir á síðustu áratuga hafa leitt í ljós verulega aukna hættu á kransæðasjúkdómi og ótímabærum dauðsföllum af þeim sökum hjá einstaklingum með arfbundna kólesterólhækkun (5-10). Tafla I sýnir samantekt úr nokkrum þessara rannsókna þar sem hættan kemur vel í ljós. Athuga ber að þessar tölur byggjast á niðurstöðum áður en ýmis meðferðarúrræði eins og statínlyf komu til sögunnar.Í Bretlandi hefur upplýsingum um einstaklinga með arfbundna kólesterólhækkun verið safnað af Simon Broome Familial Hyperlipidaemia Register frá 1980 (11). Þetta gagnasafn hefur að geyma skrár sjúklinga með arfbundna kólesterólhækkun frá 21 blóðfitugöngudeild í Bretlandi. Árið 1991 birtist uppgjör frá Simone Broome Register á hlutfallslegri áhættu á andláti af völdum kransæðasjúkdóms hjá einstaklingum með arfbundna kólesterólhækkun en fæstir voru meðhöndlaðir með kólesterólækkandi lyfjum fyrir þann tíma. Reiknuð var hlutfallslega hættan, það er hlutfall nýgengis andláta af völdum kransæðasjúkdóms hjá rannsóknarhópnum og nýgengis hjá almennu þýði. Hjá einstaklingum milli tvítugs og fertugs var hlutfallslega hættan 84 (11) sem þýðir áttatíu og fjórfalda hættu í þessum aldurshópi. Árið 1999 birti Simone Broome Register nýtt uppgjör sem sýndi að hlutfallsleg hætta þessa hóps á andláti af völdum kransæðasjúkdóms lækkaði úr 84 í 17,5 (12). Líklega má þakka þessa lækkun bættri meðferð en 86% sjúklinga í Simon Broome Register voru meðhöndlaðir með statínum í seinna uppgjörinu en mjög fáir höfðu fengið þá meðferð í fyrra uppgjörinu (12). Í eldri aldurshópum dró úr hlutfallslegu áhættunni vegna aukinnar tíðni hjartasjúkdóma í almennu þýði með vaxandi aldri. Hættan var þó enn veruleg í elsta aldurshópnum.
Niðurstöður rannsókna á arfbundinni kólesterólhækkun eru því samhljóða í að lýsa verulega aukinni áhættu á kransæðasjúkdómi hjá einstaklingum með sjúkdóminn.
Klínísk skilmerki arfbundinnar kólesterólhækkunar
Klínísk greining arfbundinnar kólesterólhækkunar getur verið vandasöm þar sem kólesterólgildi einstaklinga með erfðagallann geta verið svipuð kólesterólgildum einstaklinga sem hafa hátt kólesteról af öðrum orsökum. Kólesterólútfellingar í sinar (mynd 1) eru þó taldar nær öruggt klínískt merki um arfbundna kólesterólhækkun en næmi þess er ekki mikið þar sem aðeins helmingur einstaklinga um sextugt með sjúkdóminn hefur þessar útfellingar (1). Klíníska sjúkdómsgreiningin byggir því á kólesterólmælingu einstaklings og nánustu ættingja. Simon Broome FH Register í Bretlandi hefur sett fram eftirfarandi skilmerki:1. Heildarkólesteról hærra en 6,7 mmól/L hjá einstaklingum yngri en 16 ára en hærra en 7,5 mmól/L hjá einstaklingum eldri en 16 ára.
2. Kólesterólútfellingar í sinar viðkomandi einstaklings eða hjá fyrstu eða annarrar gráðu ættingja.
3. Ættarsaga, það er fyrstu gráðu ættingi með kransæðastíflu fyrir sextugt eða annarrar gráðu ættingi með sögu um kransæðastíflu fyrir fimmtugt. (Fyrstu gráðu ættingjar eru systkini, börn og foreldrar en annarrar gráðu ættingjar eru afar, ömmur og systkini foreldra.)
4. Fjölskyldusaga um heildarkólesteról hærra en 7,5 mmól/L í fyrstu eða annarrar gráðu ættingja.
Sjúkdómsgreining er talin örugg ef skilmerki 1 og 2 eru uppfyllt en líkleg ef skilmerki 1 og 3 eða 1 og 4 eru uppfyllt (11).
Þríglýseríðar og HDL (high density lipoprotein, háþéttnifituprótín) -kólesteról eru hins vegar eðlileg hjá einstaklingum með arfbundna kólesterólhækkun nema aðrir erfðaþættir eða umhverfisþættir komi til viðbótar.
Erlendar rannsóknir hafa sýnt að einstaklingar með arfbundna kólesterólhækkun eru iðulega ekki meðhöndlaðir á fullnægjandi hátt (13). Því er í skýrslu Alþjóðaheilbrigðisstofnunarinnar frá 1997 um arfbundna kólesterólhækkun (1) hvatt til þess að skýrar leiðbeiningar séu kynntar um greiningu og meðferð á sjúkdómnum og meðferðarúrræði á hverjum stað gerð aðgengileg. Hjartavernd leggur til eftirfarandi vinnutilhögun ef klínískur grunur um arfbundna kólesteról vaknar:
1. Auk heildarkólesteróls verði mæld þríglýseríð, háþéttnifituprótín, kreatínin, blóðsykur og TSH (thyroid stimulating hormone) í sermi eftir næturföstu.
2. Farið verði yfir klínísk skilmerki sem lýst er hér að ofan.
3. Meðferðarlæknirinn feli Hjartavernd að gera DNA-próf til að kanna hvort sjúklingurinn hafi þekkta stökkbreytingu sem orsök sjúkdómsins og hvort viðkomandi ættrekist til þekktra ætta með arfbundna kólesterólhækkun. Hjartavernd býður sjúklingum þessa rannsókn, þeim að kostnaðarlausu.
4. Hjartavernd mun senda svarbréf til viðkomandi læknis með niðurstöðum rannsóknanna.
5. Læknar geta ávallt vísað einstaklingum með háa blóðfitu á göngudeild háþrýstings og blóðfitu til greiningar og meðferðar.
Meðferð hækkaðs kólesteróls hjá einstaklingum með arfbundna kólesterólhækkun
Hornsteinn meðferðar við arfbundinni kólesterólhækkun eru lyf í flokki HMG-coA reduktasa hemlara eða statín. Þessi lyf eru mjög virk til lækkunar á LDL kólesteróli hjá einstaklingum með arfbundna kólesterólhækkun (14) og hafa rannsóknir sýnt að meðferðin dregur úr kransæðaþrengslum hjá einstaklingum í þessum sjúklingahópi (15). Rannsóknir á gildi þessarar meðferðar, bæði meðal sjúklinga með kransæðasjúkdóm og í almennu þýði, hafa einnig sýnt fram á verulegan ávinning bæði með tilliti til ævilengdar og til að hindra kransæðastíflu (16,17). Matarráðgjöf er mikilvæg en árangur hennar er minni en hjá öðrum með sambærileg kólesterógildi vegna erfðagallans.Fullorðinn einstaklingur með arfbundna kólesterólhækkun er í meiri hættu á að fá kransæðasjúkdóm en jafnaldri hans með sama kólesterólgildi og aðra svipaða áhættuþætti. Ástæða þessa er að kólesterólhækkun einstaklinga með arfbundna kólesterólhækkun er fyrir hendi frá fæðingu og börn með arfbundna kólesterólhækkun hafa einnig verulega hækkað kólesteról. Sjúkdómsgreiningin felur því í sér þörf á ákveðnari meðferð við áhættuþáttum kransæðasjúkdóms hjá einstaklingum með arfbundna kólesterólhækkun heldur en jafnöldrum með svipaða áhættuþætti að öðru leyti.
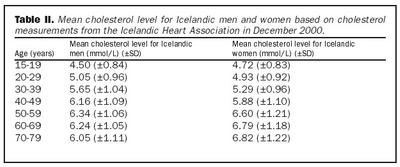
Markmið blóðfitumeðferðarinnar er að ná kólesterólgildi viðkomandi niður fyrir aldursbundið meðaltal. Í töflu II er meðaltalskólesterólgildi Íslendinga samkvæmt hóprannsókn Hjartaverndar eftir aldurshópum og kyni. Hafi viðkomandi þegar einkenni kransæðasjúkdóms er stefnt að því að koma kólesteróli vel niður fyrir 5 mmól/L. Byrja skal meðferð með statínum samkvæmt leiðbeiningum í Sérlyfjaskrá. Skammtur skal aukinn á þriggja mánaða fresti uns meðferðarmarkmiði er náð eða aukaverkanir koma fram. Oft þarf að gefa hámarksskammt af statínum og í vissum tilfellum einnig önnur blóðfitulækkandi lyf eins og fíbröt og gallsýrubindandi resín. Mikilvægt er að einstaklingar með arfbundna kólesterólhækkun reyki ekki og að háþrýstingur og aðrir áhættuþættir kransæðasjúkdóms séu vel meðhöndlaðir.
Hefja skal meðferð með statínum um 20 ára aldur en mælt er með að börnum með arfbundna kólesterólhækkun sé vísað til sérfræðings í efnaskiptasjúkdómum barna eða á göngudeild háþrýstings og blóðfitu á Landspítala Hringbraut. Statínlyfin eru ekki skráð fyrir börn og unglinga en vert er að hefja meðferð með gallsýrubindandi resínum ef kólesteról er mjög hátt.
Arfbundin kólesterólhækkun á Íslandi
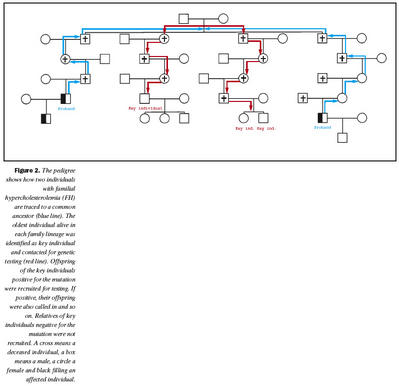
Undanfarna tvo áratugi hefur verið leitað að einstaklingum með arfbundna kólesterólhækkun á göngudeild háþrýstings og blóðfitu. Fyrir fjórum árum var hafin vinna við að þróa aðferð til að fjölga nýgreiningum á arfbundinni kólesterólhækkun á deildinni með það að lokamarkmiði að greina alla einstaklinga með arfbundna kólesterólhækkun á Íslandi. Gerð var frumrannsókn í þeim tilgangi að kanna gildi þessarar aðferðar sem byggðist á að nýta ættrakningu og erfðatækni til að afmarka rannsóknarhóp með nægilega háa tíðni af sjúkdómnum svo skimun yrði fýsileg. Erfðafræðinefnd Háskóla Íslands annaðist ættrakningu. Niðurstöður þessarar rannsóknar eru enn óbirtar en helstu niðurstöður voru að með því að ættrekja 10 einstaklinga með arfbundna kólesterólhækkun saman í þrjár ættir og skima fyrir sjúkdómnum í ættunum, fundust 45 einstaklingar með arfbundna kólesterólhækkun af 306 sem voru prófaðir. Forfeður þessara þriggja ætta voru fæddir í byrjun 19. aldar. Algengi arfbundinnar kólesterólhækkunar í þýðinu var því um 13% sem er rúmlega 60 sinnum hærra en er áætlað í almennu þýði sem er 0,2%. Rannsóknin var takmörkuð í upphafi við elstu kynslóð í hverjum ættlegg sem unnt var að kalla í (mynd 2). Þannig var ekki leitað að arfbundinni kólesterólhækkun í afkomendum þeirra sem ekki reyndust hafa sjúkdóminn. Þegar einstaklingar í þessari elstu kynslóð greindust hins vegar með sjúkdóminn voru afkomendur þeirra einnig athugaðir. Þannig greindust 37 einstaklingar til viðbótar. Erfðarannsóknirnar til þessa hafa leitt í ljós þrjár stökkbreytingar í íslenskum ættum með arfbundna kólesterólhækkun en yfir 600 stökkbreytingar hafa greinst hjá öðrum þjóðum (18). Flestar stökkbreytingar sem hafa greinst eru bundnar við ákveðnar ættir eða þjóðir. Algengasta stökkbreytingin í Íslendingum (I4T+C) er staðsett á mótum fjórðu innraðar og fjórðu útraðar og orsakar truflun í samtengingu á útröð 4 í LDL viðtakageninu (19). Þessi stökkbreyting hefur fundist hjá nokkrum einstaklingum í Bandaríkjunum og verið er að kanna hvort þeir einstaklingar eigi rætur að rekja til Íslands. Þessi stökkbreyting hefur ekki fundist á Norðurlöndum þrátt fyrir víðtæka leit. Önnur stökkbreyting (A519T) er staðsett í útröð 11 (18). Þessi stökkbreyting hefur líka fundist í Þýskalandi en ekki er vitað um tengsl Þjóðverja og Íslendinga með þessa stökkbreytingu. Þriðja stökkbreytingin er 2 kb brottfall úr útröð 9 og 10 auk innraðar 10 (20) og hefur þessi stökkbreyting aðeins fundist hér á landi. Allar valda þessar stökkbreytingar truflun á myndun LDL viðtakans og þær eru greinanlegar með aðferðum sem byggja á keðjuverkun með liðunarensími (polymerase chain reaction, PCR), sem er forsenda kerfisbundinnar leitar að þeim.
Alls hafa 206 einstaklingar greinst með arfbundna kólesterólhækkun á göngudeild háþrýstings og blóðfitu. Áætlað er að rúmlega 500 manns (1:500) á Íslandi hafi sjúkdóminn og hefur því tæplega helmingur einstaklinga með arfbundna kólesterólhækkun á Íslandi verið greindur.
Af þeim sem þegar hafa greinst með arfbundna kólesterólhækkun eru 62% (128 einstaklingar) með þekkta stökkbreytingu í geni LDL viðtakans. Algengasta stökkbreytingin (I4T+C) var áætluð valda um 60% af arfbundinni kólesterólhækkun í landinu en það mat byggðist á rannsókn á hópi óskyldra sjúklinga með arfbundna kólesterólhækkun (19). Nú hafa greinst 93 einstaklingar (43%) með þessa stökkbreytingu en 25 (11%) í útröð 11 og 10 (5%) með brottfall úr útröð 9 og 10.
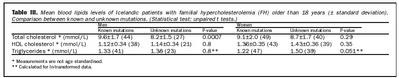
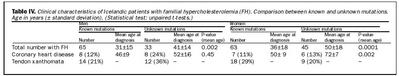
Meðalkólesterólgildi karla með þekkta stökkbreytingu var 9,6 mmól/L við greiningu. Kólesterólgildi karla var marktækt lægra hjá þeim sem hafa óþekkta stökkbreytingu eða 8,2 mmó/L (tafla III). Hjá konum var meðaltal kólesteróls sjúklinga með þekkta stökkbreytingu 9,1 mmól/L en 8,7 mmól/L hjá sjúklingum með óþekkta stökkbreytingu (P = 0,29) (tafla III). Meðalaldur við greiningu var marktækt lægri bæði hjá körlum og konum hjá þeim sem höfðu þekkta stökkbreytingu (tafla IV) sem endurspeglar væntanlega ákafari leit í þeim ættum þar sem stökkbreyting er þekkt enda greining auðveldari einkum hjá börnum og unglingum.
Um 11% kvenna með arfbundna kólesterólhækkun í þessu uppgjöri höfðu fengið kransæðastíflu (tafla IV). Konur með þekkta stökkbreytingu fengu kransæðastíflu fyrr eða um fimmtugt en konur með óþekkta stökkbreytingu um sjötugt. Þessi munur sést ekki hjá körlum. Kólesterólútfellingar í sinar fundust hjá um 20-30% einstaklinga með arfbundna kólesterólhækkun en þær eru einkennandi fyrir sjúkdóminn eins og áður var getið.
Rétt er að álykta varlega út frá frá þessum niðurstöðum þar sem um tiltölulega fáa einstaklinga er að ræða. Þó má segja að einstaklingar með þekkta stökkbreytingu hafi dæmigerð klínísk einkenni arfbundinnar kólesterólhækkunar með háu kólesteróli og kransæðasjúkdómi snemma á ævinni. Tíðni kransæðasjúkdóms er þó lág í hópnum sem líklega má rekja til þess hve lágur meðalaldur var við greiningu (30 ár hjá körlum og 37 ár hjá konum). Einstaklingar með óþekkta stökkbreytingu virðast hafa vægari klíníska mynd þar sem kólesterólgildið við greiningu er heldur lægra og kransæðasjúkdómur virðist koma seinna fram hjá þessum hópi kvenna. Því er mögulegt að mismunandi stökkbreytingar hafi í för með sér mismunandi röskun á virkni LDL viðtakans eins og erlendar rannsóknir hafa bent til (21). Einnig er mögulegt að stökkbreytingar í öðrum genum eins og apólípóprótín B geninu eða í nýuppgötvuðu geni á litningi 1 (22) sé orsök klínísku myndarinnar. Enginn þessara einstaklinga hefur þó greinst með stökkbreytinguna apólípóprótín B 3500 samkvæmt birtum og óbirtum niðurstöðum höfunda (23).
Framtíðaráform
Í þessari grein hefur verið rætt allítarlega um mikilvægi þess að greina og meðhöndla einstaklinga með arfbundna kólesterólhækkun. Niðurstöður frumrannsóknar sem höfundar stóðu að sýndu fram á mikinn ávinning af skimun fyrir arfbundinni kólesterólhækkun í íslenskum ættum og var það kveikjan að því átaki sem nú er hafið hjá Hjartavernd. Gagnagrunnur Hjartaverndar verður nýttur til að finna einstaklinga með mjög hátt kólesteról og ættrekja þá til sameiginlegra forfeðra. Einnig verður kannað hvort þeir tengjast ættum sem þegar eru þekktar á göngudeild háþrýstings og blóðfitu. Vonast er til að með þessum hætti verði skilgreint þýði með háa tíðni sjúkdómsins þar sem skimun skilar miklum ávinningi.
Hver nýr einstaklingur sem finnst með arfbundna kólesterólhækkun er mikilvægur, ekki aðeins vegna þess einstaklings heldur einnig vegna þeirra nákomnu ættingja sem oftast greinast einnig þegar ný ætt hefur verið skilgreind og skimun hafin. Hjartavernd mun framkvæma sjúkdómsgreinandi erfðapróf og kanna ættartengsl við þekktar ættir hjá þátttakendum í átaki því sem nú er hafið. Það er von höfunda að þeir læknar sem telja að skjólstæðingar þeirra hafi arfbundna kólesterólhækkun beini þeim til rannsóknar og leggi því lið að fleiri greinist með sjúkdóminn.
Þakkir
Höfundar vilja þakka starfsfólki Hjartaverndar og starfsfólki göngudeildar háþrýstings og blóðfitu Landspítala Hringbraut fyrir margvíslega aðstoð við gerð þessarar greinar.Heimildir
1. Familial hypercholesterolemia (FH) a WHO Report, 3 October 1997. Geneva: WHO; 1997: 1-45.2. Goldstein JL, Sobhani MK, Faust JR, Brown MS. Heterozygous familial hypercholesterolemia: failure of normal allele to compensate for mutant allele at a regulated genetic locus. Cell 1976; 9: 195-203.
3. Goldstein JL, Hobbs HH, Brown MS. Familial Hypercholesterolemia. In: Scriver C, ed. The Metabolic Bases of Inherited Diseases. 7th ed. New York: McGraw-Hill Companies; 1994: 1981-2029.
4. Goldstein JL, Schrott HG, Hazzard WR, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest 1973; 52: 1544-68.
5. Mabuchi H, Miyamoto S, Ueda K, Oota M, Takegoshi T, Wakasugi T, et al. Causes of death in patients with familial hypercholesterolemia. Atherosclerosis 1986; 61: 1-6.
6. Slack J. Risks of ischaemic heart-disease in familial hyperlipoproteinaemic states. Lancet 1969; 2: 1380-2.
7. Jensen J, Blankenhorn DH, Kornerup V. Coronary disease in familial hypercholesterolemia. Circulation 1967; 36: 77-82.
8. Heiberg A. The risk of atherosclerotic vascular disease in subjects with xanthomatosis. Acta Med Scand 1975; 198: 249-61.
9. Stone NJ, Levy RI, Fredrickson DS, Verter J. Coronary artery disease in 116 kindred with familial type II hyperlipoproteinemia. Circulation 1974; 49: 476-88.
10. Beaumont V, Jacotot B, Beaumont JL. Ischaemic disease in men and women with familial hypercholesterolaemia and xanthomatosis. A comparative study of genetic and environmental factors in 274 heterozygous cases. Atherosclerosis 1976; 24: 441-50.
11. Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ 1991; 303: 893-6.
12. Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management. Scientific Steering Committee on behalf of the Simon Broome Register Group. Atherosclerosis 1999; 142: 105-12.
13. Williams RR, Schumacher MC, Barlow GK, Hunt SC, Ware JL, Pratt M, et al. Documented need for more effective diagnosis and treatment of familial hypercholesterolemia according to data from 502 heterozygotes in Utah. Am J Cardiol 1993; 72: 18D-24D.
14. Havel RJ, Hunninghake DB, Illingworth DR, Lees RS, Stein EA, Tobert JA, et al. Lovastatin (mevinolin) in the treatment of heterozygous familial hypercholesterolemia. A multicenter study. Ann Intern Med 1987; 107: 609-15.
15. Kane JP, Malloy MJ, Ports TA, Phillips NR, Diehl JC, Havel RJ. Regression of coronary atherosclerosis during treatment of familial hypercholesterolemia with combined drug regimens. JAMA 1990; 264: 3007-12.
16. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344: 1383-9.
17. Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med 1995; 333: 1301-7.
18. The low density lipoprotein receptor (LDLR) gene in familial hypercholesterolemia. http://www.ucl.ac.uk/fh/
19. Gudnason V, Sigurdsson G, Nissen H, Humphries SE. Common founder mutation in the LDL receptor gene causing familial hypercholesterolaemia in the Icelandic population. Hum Mutat 1997; 10: 36-44.
20. Taylor R, Bryant J, Gudnason V, Sigurdsson G, Humphries S. A study of familial hypercholesterolaemia in Iceland using RFLPs. J Med Genet 1989; 26: 494-8.
21. Kotze MJ, De Villiers WJ, Steyn K, Kriek JA, Marais AD, Langenhoven E, et al. Phenotypic variation among familial hypercholesterolemics heterozygous for either one of two Afrikaner founder LDL receptor mutations. Arterioscler Thromb 1993; 13: 1460-8.
22. Hunt SC, Hopkins PN, Bulka K, McDermott MT, Thorne TL, Wardell BB, et al. Genetic localization to chromosome 1p32 of the third locus for familial hypercholesterolemia in a Utah kindred. Arterioscler Thromb Vasc Biol 2000; 20: 1089-93.
23. Guðnason V, Sigurðsson G, Humphries S. Notkun ensímhvattrar fjölföldunar á DNA til að skima eftir erfðagalla í apóprótíni-B í íslenskum fjölskyldum med hátt kólesteról í sermi. Læknablaðið 1990; 76: 431-6.