
07/08. tbl. 94.árg. 2008
Fræðigrein
Creutzfeldt-Jakob sjúkdómur og riða í sauðfé
Ágrip
Tilgangur: Sauðfjárriða og Creutzfeldt-Jakob sjúkdómur (CJD) hjá mönnum teljast til príonsjúkdóma og smitefni þeirra eru náskyld. Leitað var svara við spurningunni hvort sauðfjárriða gæti borist í menn og valdið CJD.
Efniviður og aðferðir: Skimað var fyrir CJD á 40 ára tímabili, 1960-2000. Afturskyggn rannsókn hófst 1980. Sjúkraskrár taugasjúkdómadeildar Landspítalans frá árunum 1960-1980 voru kannaðar. Rannsakaðir voru vefjabitar (vaxkubbar) úr safni rannsóknarstofu HÍ við Barónsstíg af tilfellum, sem höfðu verið greind sem CJD eða rökstuddur grunur var um þá greiningu.Við framskyggnu rannsóknina 1980-2000 gafst einnig færi á að kanna tákna 129 í erfðaefni príons og auðkenna stofn smitefnis. Teknar voru saman upplýsingar um faraldsfræði riðu og neysluvenjur landans kannaðar.
Niðurstöður: 4 tilfelli af CJD voru greind á þessum 40 árum, tvö á hvoru tímabili. Árleg dánartíðni var 0,44 á milljón íbúa.
Ályktun: Þar eð tíðnin hérlendis er ríflega helmingi lægri en meðaltalstíðni í 18 öðrum Evrópulöndum á tímabilinu 1997-2004, sem var 1,11 á milljón íbúa, teljum við næsta víst að sauðfjárriða berist ekki í fólk, enda hefði má búast við hærri tíðni CJD hérlendis og hugsanlega afbrigðilegum tilfellum ef svo væri því Íslendingar hafa verið útsettir fyrir riðu í 130 ár.
Inngangur
Riða er frumgerð príon sjúkdóma, sem einnig eru nefndir smitandi svampkennd heilamein (transmissible sprongiform encephalopathy, TSE´s) sem dregur dám af einkennandi vefjaskemmdum í heila. Sjúkdómarnir eru banvænir hrörnunarsjúkdómar í miðtaugakerfi manna (neurodegenerative) og dýra. Helstir þeirra eru, auk riðu í sauðfé og geitum, kúariða (bovine spongiform enceph- alopathy, BSE), langvinnur rýrnunarsjúkdómur í hjartardýrum (Chronic Wasting disease, CWD). Í mönnum eru helstu sjúkdómarnir: Creutzfeldt-Jakob (CJD), Kuru og banvænt ættlægt svefnleysi (Fatal Familial Insomnia, FFI). Dýrasjúkdómarnir eru að því leyti frábrugðnir mannasjúkdómum að þeir eru að jafnaði smitandi, en hins vegar geta mannasjúkdómarnir auk þess að vera smitandi, verið ættlægir eða stakir (sporadic). Greind eru fjögur mismunandi form af CJD (sjá aðferðir) og er staka formið sCJD langalgengast eða milli 80-90% (1,2,3) Megineinkenni þess eru hratt vaxandi vitglöp (dementia) (4). Orsök er ókunn en þekktur áhættuþáttur er methionine/ methionine (M/M) í tákna 129 príon-gensins (5).
Príon (proteinacous infectious particles) er heitið sem Prusiner gaf smitefninu þegar hann setti fram þá kenningu að smitefnið væri einvörðungu gert úr prótíni og hefði ekkert erfðaefni (6). Nokkru síðar var sýnt fram á gen sem skráir fyrir eðlilegu príon-prótíni PrPC, (c=cellular). Þetta er himnutengt prótín sem er tjáð í flestum vefjum en sýnu mest í miðtaugakerfi (7). Enda þótt PrPC sé mjög vel varðveitt milli tegunda, til dæmis er 90% samsvörun með manni og mús og 97% með kind og manni, er þekking á starfi þess brota- og mótsagnakennd. Mýs með óvirku PrPCgeni (knock-out mice) sem fylgst var með í tvö ár þroskuðust eðlilega (8). En hins vegar hefur ýmist verið lýst eituráhrifum PrPC í umfrymi taugafrumu eða verndandi (9,10). Rannsóknir á tvíblendingskerfi gersveppa (yeast two-hybrid) hafa leitt í ljós að PrPC binst prótíni (neurotrophin receptor interacting MAGE homlog ) í umfrymi, og að samtjáning þeirra í taugafrumum geti valdið lækkaðri himnuspennu í hvatberum sem getur verið forboði stýrðs frumudauða (apoptosis) eða raskað flutningi taugaboða með GABA-taugafrumum (11). Smitefnið PrPSc myndast við víxlverkun við PrPC og virkar bæði sem mót fyrir PrPC og örvar tjáningu þess. Við þessi samskipti breytist þrívíddarform PrPC, í stað α-spírala myndast β-flákar (B-pleated sheets), sem gerir smitefnið mjög þolið, m.a. gegn venjulegum sótthreinsunaraðferðum. Amínosýruröðin er hins vegar hin sama og í PrPC og því greinir líkaminn smitefnið ekki sem framandi og ónæmiskerfið bregst ekki við því með mótefnamyndun (12,13). Tilvist PrPC er skilyrði fyrir því að smit eigi sér stað. Mýs, sem PrP geni hefur verið eytt (knock-out mice), voru ónæmar fyrir sýkingu með riðu (14).
Þrátt fyrir að mikil þekking hafi unnist síðan príon-kenningin var sett fram, sem hefur stutt þá kenningu (12,13,15), var það ekki fyrr en nýlega að ætla má að hún hafi verið sönnuð, þegar tókst að sýna fram á að smitefni myndað in vitro væri smitandi in vivo með tilraunasýkingu í músum (16) og hömstrum (17).
Íslendingar hafa búið við riðu í sauðfé í um það bil 130 ár. Hún kom fyrst upp í Skagafirði og breiddist fljótt út um Mið-Norðurland og var bundin við það svæði í 75 ár, en tók þá að breiðast út til annarra landsvæða (18, 19) og náði til 2/3 hluta landsins þegar hún náði hámarki um miðjan níunda áratuginn (20).
Hvatinn að þessari rannsókn á CJD hérlendis sem hófst 1980 voru greinar um háa tíðni CJD í líbýskum Gyðingum í Ísrael sem var um þrítugfalt hærri en í fólki af öðrum kynþáttum í landinu (21). Í kjölfar þess var sett fram sú tilgáta að hugsanlega mætti rekja þessa háu tíðni til neyslu augna úr sauðfé, sem telst til lostætis hjá Líbýumönnum. Það að augu eru sérstaklega tilgreind má rekja til þess að um svipað leyti var skýrt frá því að CJD hafi sennilega borist með ígræðslu hornhimnu (22). Líklegra virtist þó að smitið hafi orðið vegna neyslu þeirra á léttsteiktum heila og mænu en það þykir einnig mikið lostæti ekki einungis hjá líbýskum Gyðingum heldur einnig Aröbum af ýmsu þjóðerni í Norður-Afríku (23).
Efniviður og aðferðir
Fyrri hluti rannsóknar á tíðni CJD, sem hófst 1980, var afturskyggn og náði yfir tímabilið 1960 til 1980. Farið var yfir sjúkraskýrslur taugalækningadeildar Landspítala og leituð upp tilfelli sem höfðu ýmist verið greind sem CJD eða þar sem rökstuddur grunur var um að sjúklingur hefði látist af CJD. Jafnframt voru fengnir vefjabitar úr safni rannsóknarstofu Háskólans við Barónsstíg. Alls fundust 5-10 paraffín-kubbar úr heila úr hverju tilfelli. Nýjar sneiðar voru skornar og litaðar með haematoxylin eosin (HE) , anti-GFAP (glial fibriallary acidic protein) fyrir stjarnfrumum og með mótefni gegn PrPSc. Síðari hluti rannsóknarinnar, það er frá 1980 til 2000, var framskyggn. Við krufningu voru áður en þeir voru hertir í formalíni, teknir bitar og snöggfrystir í fljótandi köfnunarefni sem gerði kleift að beita öðrum rannsóknaraðferðum en vefjameinafræðilegum. Sýni fyrir smásjárskoðun voru tekin úr: frontal, parietal, temporal og occipital cortex, basal ganglia, thalamus með substantia nigra, mesencephalon, hippocampus, cerebellum og medulla oblongata.
Við klíníska greiningu á CJD var stuðst við eftirfarandi skilmerki sem notuð voru af samstarfshópi Evrópusambandsins (NEUROCJD) sem fékkst við skimun og rannsókn á CJD í Evrópu undir forystu Roberts G. Will í Edinborg og við tókum þátt í.
Meinvefjafræði
Skornar voru 5μm þykkar sneiðar og þær litaðar með HE litun til að meta einkennandi vefja-skemmdir, en jafnframt ónæmislitaðar til að sýna fram á smitefnið PrPSc og viðbrögð stjarnfrumna. Sams konar athugun var gerð á vefjasneiðum úr riðu til samanburðar.
Við ónæmislitun notuðum við peroxidase-antiperoxidase (PAP) aðferð með diaminobenzidine (DAB) sem hvarfefni. Við litun fyrir PrPSc beittum við formeðhöndlun samkvæmt (25), og 3F4 einstofna músamótefni gegn PrPSc(Covance, Leeds, UK) eða 3F10(Cayman Chemical, Ann Arbor, Michigan, USA) í þynningunni 1:2000. En til að sýna fram á smitefnið í sauðfé beittum við einstofna mótefninu F89/160.1.5 í þynningunni 1:3000 (26). Við litun fyrir stjarnfrumum notuðum við viðurkenndar aðferðir (27). Þar beittum við fjölstofna kanínumótefni gegn Glial Fibrillary Acidic Protein (anti-GFAP, Dakocytomation, Glostrup, Danmörk) í þynningunni 1:250. GFAP er einkennandi fyrir þráðlaga stjarnfrumur (fibrillary astrocytes) í hvítu. Í gráma, það er heilberki og kjörnum, eru hins vegar gemistocytic stjarnfrumur sem litast ekki með mótefni gegn GFAP, en bregðast við áreiti með aukinni myndun af glial fibrillary acidic prótíni og taka á sig form þráðlaga stjarnfrumna.

Paraffinkubbar úr þessum tilfellum voru sendir til Herberts Budka í Vín vegna ESB- verkefnis, Prionet, sem hann stýrir og við tókum þátt í.
Niðurstöður
Í afturskyggnu rannsókninni sem náði yfir 20 ár (1960-1980) fundust tvö tilfelli af stökum Creutzfeldt-Jakob sjúkdómi (sCJD).
Fyrsta tilfellið: 56 ára verkamaður. Utan þunglyndis um hríð hafði hann verið hraustur. Um það bil 4½ fyrir andlátið fór að bera á minnisleysi og nokkru síðar kom fram stirðleiki, magnleysi og ofskynjanir. Hann varð óvinnufær og var lagður inn á sjúkrahús á landsbyggðinni 1½ mánuði eftir að fyrstu einkennin komu fram. Einkennin fóru sífellt versnandi og var hann fluttur á Kleppsspítalann eftir 1½ mánuð með sjúkdómsgreininguna geðklofi. Gunnar Guðmundsson rannsakaði hann þar og taldi samkvæmt klínískri skoðun að um CJD væri að ræða. Auk stigvaxandi vitglapa komu fram sjóntruflanir, vöðvakippir (myoclonus), skortur á samhæfingu (coordination) og pyramidal og extrapyramidal einkenni, að lokum lá hann hreyfingarlaus og gat ekki tjáð sig. Heilarit var dæmigert fyrir sCJD. Engin ættarsaga um geðsjúkdóma.
Endanleg klínísk greining: líklegur CJD
Gerð var krufning og við vefjaskoðun sáust einkennandi svampkenndar (spongiform) breytingar í sneiðum úr heilaberki, fjölgun og stækkun stjarnfrumna og með ónæmislitun fundust útfellingar af PrPSc ýmist sem taugamóta (synaptic) eða perivakuólar. Ekki sáust flákar (plaques).
Endanleg greining: ótvíræður sCJD
Annað tilfellið: 73 ára bóndi. Fæddur og uppalinn og bjó á svæði sem riða hefur aldrei fundist. Hafði verið heilsugóður, en 5½ mánuðum fyrir andlátið fór hann að verða undarlegur í háttum, sljór og gleyminn og átti erfitt með að tjá sig. Gunnar Guðmundsson skoðaði sjúklinginn fyrir innlögn á Landspítalann og þar lést hann eftir ríflega fjögurra mánaða legu. Hann var ruglaður við innlögn og gat ekki svarað spurningum og fóru einkennin síversnandi. Megineinkenni voru vaxandi vitglöp og kippir í útlimum. Auk þess var hann með sjóntruflanir, einkenni frá litla heila og pyramidal og extrapyramidal einkenni. Að lokum var hann rúmliggjandi og tjáði sig ekki. Heilarit sýndi dæmigerðar breytingar fyrir CJD. Klíníska greiningin var vitglöp, sennilega vegna CJD. Sjúklingur andaðist 5½ mánuði eftir upphaf einkenna.
Endanleg klínísk greining: líklegur sCJD
Hann var krufinn og reyndist dánarorsök lungnabólga. Lýst var heilarýrnun með víkkun á heilahólfum.
Smásjárskoðun á sneiðum úr heila leiddi í ljós dæmigerðar svampkenndar vefjaskemmdir og jafnframt tókst með ónæmislitun að sýna fram á smitefnið PrPSc og voru útfellingar af svonefndu synaptic formi í heilaberki og berki litla heila, en ekki sáust flákar. Fjölgun og stækkun stjarnfrumna.
Endanleg greining: ótvíræður sCJD
Árleg dánartíðni á þessu 20 ára tímabili sem afturskyggna rannsóknin náði til reyndist vera 0,49 á milljón íbúa.
Framskyggn rannsókn frá 1980 til 2000
Þriðja tilfellið: 72 ára húsmóðir, borin og barnfædd í Reykjavík og bjó þar alla ævi. Þegar hún var fimmtug var græddur í hana hjartagangráður. Einnig hafði verið greind hjá henni vanstarfsemi skjaldkirtils og hafði hún fengið lyfjameðferð vegna hennar.
Að öðru leyti hafði hún verið hraust þar til einkenni þess sjúkdóms komu í ljós sem leiddi til innlagnar á sjúkrahús. Fyrstu einkennin voru minnisleysi og breyting á hegðun. Hún svaraði spurningum útí hött. Síðar fór að bera á óstöðugleika og dettni. Síðan komu fram einkenni vaxandi vitglapa, sjóntruflanir, einkenni frá litla heila og pyramidal einkenni og loks var hún alveg rúmliggjandi og ófær um að tjá sig. Heilarit sýndi breytingar dæmigerðar fyrir CJD. Sjúklingur lést um það bil þremur mánuðum eftir að fyrstu einkenni komu fram. Við krufningu var heilinn næsta eðlilegur við stórsæja skoðun. Sýni voru tekin í formalín fyrir smásjárskoðun úr heilaberki, frontalt, parietalt, temporalt og occipitalt, basalganglia, corpora mamillaria, thalamus, substantia nigra, pons með locus coeruleus, pes hippocampi og cerebellum. Auk þess var tekinn biti og snöggfrystur í fljótandi köfnunarefni fyrir aðrar rannsóknir. Engin ættarsaga um geðsjúkdóma.
Klínísk greining: líklegur sCJD
Við smásjárskoðun á sneiðum úr heila sáust mjög vægar svampkenndar breytingar í heilaberki frontalt og parietalt, en mjög áberandi og útbreiddar í temporal, occipital og parahippocampal berki. Mjög vægar breytingar í substantia nigra og molecular lagi litla heila. Með ónæmislitun fundust útfellingar af PrPSc synaptic og perivacuólar, ríkulegt í heilaberki en minna áberandi í berki litla heila. Ekki sáust flákar. Fjölgun og stækkun stjarnfrumna var ótvíræð.
Endanleg greining: ótvíræður sCJD
Aðrar rannsóknir: Ekki fundust stökkbreytingar í PrP geni; Tákni 129 í PrP geni reyndist arfhreinn methionine/methionine (M/M).Við athugun með prótínþrykki á PrPSc reyndist smitefnið vera af flokki 1, sem er einkennandi fyrir sCJD.
Fjórða tilfellið: 70 ára húsmóðir borin og barnfædd í Reykjavík og bjó þar alla tíð. Hún hafði verið heilsuhraust og ekki legið á sjúkrahúsi nema við barnsburð. Ári áður en hún lést fór að bera á óöryggi í stöðu og við göngu, það er slingri. Þau einkenni ágerðust og jafnframt dró úr þoli. Hún varð að hætta störfum fjórum mánuðum eftir að einkennin komu fram og var lögð inn á sjúkrahús tveimur mánuðum síðar. Við komu var hún mjög stíf og talsvert bar á handskjálfta. Segulómmynd (MRI) tekin ríflega mánuði eftir innlögn sýndi lítinn blett í frontal cortex vinstra megin, sem ekki hafði sést fjórum mánuðum áður. Endurtekin heilarit voru ekki dæmigerð fyrir CJD. Um svipað leyti voru greind vitglöp með áberandi frontal einkennum. Þessi einkenni fóru síversnandi og síðasta mánuðinn sem hún lifði lá hún að mestu hreyfingarlaus og gat ekki tjáð sig. Engin ættarsaga. Einkenni voru talin benda til corticostriatonigral degenerationar, en einnig var hugleidd greiningin CJD.
Við krufningu á heila sást lítils háttar rýrnun á frontal cortex með vægri víkkun á frontal hluta hliðarhólfa. Caput nuclei caudati virtust lítið eitt útflattir, sennilega vegna útvíkkunar á heilahólfum frekar en rýrnun.
Klínísk greining: hugsanlegur sCJD
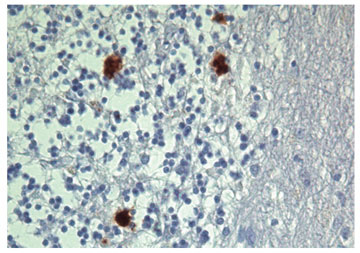
Mynd 1. Cerebellum með nokkrum mýlildisflákum í granule frumulagi, þar sem nokkur rýrnun sést. Mótefnalitun með 3F4 gegn PrPSc og haematoxylin mótlitun.
Smásjárskoðun á heila leiddi í ljós útbreiddar svampkenndar breytingar í heilaberki einkum frontalt, parietalt og temporalt en mjög vægar occipitalt. Að auki voru mjög áberandi svampkenndar breytingar í corpus striatum og thalamus, en heldur vægari í mesencephalon og medulla oblongata og þar einkum í nucleus olivarius. Væg depigmentatio í substantia nigra. Í litla heila sáust svampkenndar breytingar einkum í nucleus dentatus og granule frumulagi. Við ónæmislitun fyrir PrPSc sáust einstaka flákar í heilaberki en allmargir í litla heila einkum í granule frumulagi (mynd 1). Að auki sáust kornóttar perivakuólar útfellingar svo og í umfrymi einstakra taugafrumna og í taugamótum. Einnig sást fjölgun og stækkun stjarnfrumna. Aðrar rannsóknir leiddu í ljós að tákni 129 í PrP geni var arfblendinn methionine/valine (M/V). Með prótínþrykki var sýnt fram á að PrPSc var af flokki 1.
Endanleg greining: ótvíræður sCJD
Þessi tvö tilfelli frá 20 ára tímabili framskyggnu rannsóknarinnar svara til nýgengis 0,40 tilfelli á milljón íbúa á ári. Ef tíðnin er tekin saman fyrir allt tímabilið, það er fjögur tilfelli á 40 árum, reiknast tíðnin vera 0,44 á milljón íbúa. Við staðtölulega útreikninga var forritið SAS/STAT útgáfa 8.1 notað.
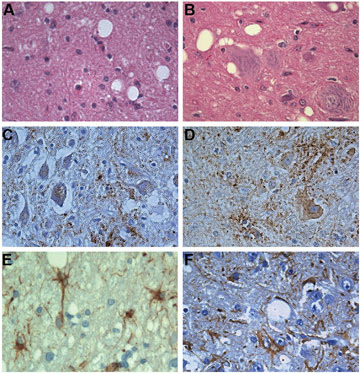
Mynd 2. A og B: Einkennandi svampkenndar (spongiform) breytingar í heilaberki í CJD (A) og í nucleus facialis í mænukylfu í riðu (B) HE; C og D: Útfellingar af PrPSc í nucleus dentatus í CJD (C) og í nucleus facialis í mænukylfu í riðu (D), sem eru sambærilegar, það er á taugamótum (synaptic) í neuropil og einnig í frymi taugafrumna. Ónæmislitun fyrir PrPSc og mótlitun með haematoxylin. E og F: Viðbrögð stjarnfrumna í heilaberki í CJD (E) og í nucleus facialis í mænukylfu í riðu (F). Ónæmislitun fyrir GFAP og mótlitun með haematoxylin.
Megindrættir í vefjaskemmdum í riðu voru sambærilegir við það sem einkennir vefjaskemmdir í CJD, það er svampkenndar breytingar í gráma og jafnframt áberandi fjölgun stjarnfrumna bæði í gráma og hvítu. Kornóttar útfellingar af PrPScí neuropil og frymi taugafrumna var áberandi (mynd 2). Mýlildisflákar (amyloid plaques) voru mjög sjaldséðir.
Umræða
Öll tilfellin, sem við greindum voru sCJD, sem er algengasti príonsjúkdómur manna. Rannsóknir á CJD innan ramma Evrópusambandsverkefnis NEUROCJD sem náði til 10 Evrópuríkja og Ísraels á árunum 1997-2004 leiddi í ljós að af 1320 dauðsföllum voru 78,6% vegna sCJD, 9% vegna gCJD, 2,1% vegna iCJD og 10,3% vegna vCJD (1). Í fyrri rannsókn á CJD í sex Evrópulöndum reyndist hlutfall sCJD nokkru hærra, 87% (2) og sama gildir um umfangsmikla samantekt á dauðsföllum vegna CJD í Evrópu, Ástralíu og Kanada, eða 83,7% (3).
Fyrstu einkennin í þremur af okkar tilfellum voru hratt vaxandi vitglöp, en í einu tilviki voru upphafseinkennin frá litla heila. Þetta kemur vel heim við niðurstöður úr NEUROCJD rannsókninni, þar sem fyrstu einkenni voru hratt vaxandi vitglöp (74%) og slingur (9%) (1). Og er það í samræmi við fleiri rannsóknir (4, 5).
Eins og fyrr getur er sjúkdómurinn ætíð banvænn og leiðir til dauða að jafnaði á 8 (4) eða 5 mánuðum (5) að meðaltali, en með talsverðum breytileika allt frá einum mánuði til 130 mánaða. Í okkar rannsókn lifðu sjúklingarnir að meðaltali í 6,25 mánuði.
CJD sem hefst með slingri, eins og fjórða tilfelli okkar, er kennt við Brownwell og Oppenheimer, sem fyrstir lýstu þeirri sýnd (28). En það gildir einungis ef vitglöp koma ekki fram fyrr en að minnsta kosti mánuði síðar (29). Í okkar tilfelli liðu um það bil sex mánuðir þar til vitglöp komu fram. Við afturskyggna rannsókn á 618 sCJD tilfellum sem höfðu verið staðfest með krufningu í Bretlandi fundust 29 slík tilfelli á 15 ára tímabili, það er 5% (29). Klínísk greining vafðist mjög fyrir mönnum og voru helstu mismunagreiningar, efnaskiptasjúkdómar, paraneoplastic heilkenni og afmýlandi sjúkdómar. Helstu byrjunareinkenni voru slingur (69%), svimi (21%), léleg samhæfing (7%) og þvoglumæli (3%), sem kemur nokkuð vel heim við okkar tilfelli. Sjúkdómurinn varaði frá 2 upp í 36 mánuði, að meðaltali 9 mánuði, sem er nokkuð skemmri tími en í okkar tilfelli, en lengri en meðaltal af öðrum tilfellum af sCJD. Einkennandi heilarit var aðeins í 10% tilfellanna.
Breytileiki (polymorphism) í tákna 129 PrP gensins, sem hefur áhrif á svipfar (phenotype) sCJD hefur verið kannaður og reyndist algengasti breytileikinn í tákna 129 vera M/V, sem tengist lengra sjúkdómsferli (30). Útfellingar af PrPSc í formi mýlildisfláka koma einkum fram í sCJD sjúklingum sem eru með samsætuna valine (allele) í tákna 129 annaðhvort sem V/V eða M/V (31).
Dánaraldur okkar tilfella var að meðaltali 68 ár. Dánartíðni er mest á aldrinum 60-80 ára samkvæmt ýmsum rannsóknum og lækkar mjög fyrir fimmtugt og yfir áttrætt (1- 4). Að vísu hefur verið sýnt fram á tilhneigingu til hækkunar á dánartíðni vegna sCJD í fólki eldra 75 ára við samanburð í Englandi og Wales frá 1970 til 2000, sem sennilega má rekja til betri skimunar (32). sCJD greinist sárasjaldan í fólki yngra en 45 ára. En einstaka sinnum finnast tilfelli af sCJD sem leggja ungt fólk að velli. Þannig hafa fundist tvö tilfelli í Bretlandi í skimun sem nær aftur til 1970, annað var 16 ára og hitt tvítugt. Í tilfellum af sCJD sem koma fram hjá svo ungu fólki er aðalmismunagreiningin vCJD sem kemur einkum fram hjá ungu fólki (33).
Dæmigerðar breytingar þriggja fasa reglubundinn útsláttur sjást oft í heilariti við sCJD. Þrjú af okkar tilfellum voru með dæmigert heilarit, en fjórða tilfellið ekki. Í NEUROCJD rannsókninni reyndust aðeins 44,6% heilarita dæmigerð í sCJD, en það skiptir máli í hvaða sjúkdómsfasa heilaritið er tekið og oft þarf að endurtaka það nokkrum sinnum (1). Önnur rannsóknaraðferð sem hefur sannað gildi sitt og ekki var beitt í okkar tilfellum er leit að 14-3-3 próteini í mænuvökva og hefur næmi þessar aðferðar reynst 96% í tilfellum af sCJD, sem staðfest hafa verið með smásjárskoðun á heila (24). Þessi aðferð hefur því verið tekin upp sem skilmerki fyrir klíníska greiningu. Að lokum skal getið að enn ein rannsóknaraðferð kann að koma að notum við greiningu á sCJD en það er segulómun á heila, þó að næmi hennar sé minni en greining 14-3-3 próteins í mænuvökva og hún hafi enn ekki verið tekin upp sem skilmerki fyrir klínískri greiningu (24).
Við smásjárskoðun fundum við dæmigerðar svampkenndar breytingar í heilaberki og berki litla heila, og kjörnum í heila og í litla heila. Þessar breytingar voru misjafnlega útbreiddar, mest áberandi í fjórða tilfellinu. Einnig sást fjölgun og/eða stækkun stjarnfrumna og einhver eyðing taugafrumna, sem var raunar erfitt að meta nema helst í granule frumulagi litla heila, þar sem snið úr honum lágu fyrir eins og í þriðja og fjórða tilfellinu. Þessar breytingar, það er svampkenndar breytingar í gráma, astrocytosis og eyðing taugafrumna, eru taldar einkennandi fyrir sCJD. Að auki skiptir máli fyrir greininguna form útfellinga PrPSc sem er að jafnaði dreifð synaptic og/eða í blettum umhverfis safabólur. Mýlildisflákar (amyloid plaques) eru hins vegar fremur sjaldgæfir í sCJD en mjög áberandi í gCJD (4, 34).
Í framskyggnu rannsókninni voru fryst heilasýni rannsökuð frekar í Edinborg (NEUROCJD) með próteinþrykki (Western Blot), sem nýtist til að greina að mismunandi stofna smitefnis, og greining á breytileika í tákna 129 PrP gensins, sem getur skýrt breytileika í svipfari sjúkdómsins (5). Við próteinþrykk kom í ljós að smitefnið var af flokki 1, sem staðfestir enn betur greininguna á þriðja og fjórða tilfelli okkar sem sCJD, því að 70% sCJD hafa þennan stofn smitefnis (5).
Nýgengi sCJD í afturskyggnu rannsókninni 1960-1980 (0,5 sjúklingar á milljón íbúa á ári; 95% öryggisbil 0,1-2,0) var lítið eitt hærra en í framskyggnu rannsókninni, 1980-2000 (meðaltal 0,4 per milljón íbúa per ár; 95% öryggisbil 0,1-1,6). Fyrir allt 40 ára tímabilið var meðal árlegt nýgengi 0,44 tilfelli á milljón íbúa á ári; 95 % öryggisbil 0,2-1,7). Ekki reyndist marktækur munur á fyrra 20 ára tímabilinu og seinna 20 ár tímabilinu, p=0,8247. Reiknað var með Poisson dreifingu í þessum útreikningum. Þessi tíðni er mun lægri en sú tíðni sem hefur fundist í öllum umfangsmiklum rannsóknum sem hafa verið gerðar. Í NEUROCJD rannsókninni reyndist tíðnin vera að meðaltali 1,0 á milljón íbúa. Það var nokkur breytileiki milli landa en þó innan tiltölulega þröngra marka, það er frá 0,7 í Grikklandi og Ísrael til 1,5 í Belgíu þannig að tíðnin hérlendis sker sig nokkuð úr (1). Við fengum tvö tilfelli til krufningar á síðara tímabilinu, þar sem grunur lék á að um CJD væri að ræða, þar sem krufning leiddi í ljós að í öðru tilfellinu var um Alzheimer að ræða en í hinu Parkinson sjúkdómur með byrjandi elliglöp (klínískt). Þessir sjúkdómar koma helst til greina sem mismunagreining, en CJD sker sig úr vegna hratt vaxandi vitglapa og skamms sjúkdómsgangs.
Þótt orsök sCJD sé óþekkt hefur verið sýnt fram á að M/M í tákna 129 er áhættuþáttur fyrir sCJD (1, 3, 5) og hefðum við mátt búast við sambærilegri tíðni og í öðrum Evrópulöndum, þareð rannsóknir á heilbrigðu þýði hérlendis leiddi í ljós að tíðni M/M í tákna er 46,6%, sem er með einni undantekningu ekki marktækt frábrugðinn því sem gerist í Vestur- og Suður-Evrópu (35). Við höfum ekki viðhlítandi skýringu á því hve lág tíðni sCJD er hérlendis, en hugsanlega gæti verið að verndandi breytileika sé að finna í öðrum táknum PrP gensins í Íslendingum. Í Asíu er tíðni M/M mun hærri, sem dæmi má nefna að í Japan er tíðni M/M í heilbrigðu þýði 93,2% en tíðni sCJD aðeins 0,82 í milljón íbúa og þar virðist samsætan lysine í tákna 219 vernda gegn sCJD (36).
sCJD og aðrir príonsjúkdómar eru banvænir og ekki er þekkt nein meðferð sem gagnast. Með nýlegum rannsóknum á sauðfjárriðu hafa fundist fjölmörg efni sem virðast lækna príonsýkingu in vitro en hafa ekki dugað in vivo og það gildir einnig um ónæmismeðferð. Undirstrikað er að þörf sé mun meiri grunnþekkingar, ekki hvað síst á PrPC (37).
Eins og að ofan getur hafa Íslendingar búið við riðu í sauðfé í um það bil 130 ár. Það er ljóst að þeir hafa verið útsettir fyrir riðu í verulegum mæli bæði vegna neyslu sauðfjárafurða og við hirðingu sauðfjár.
A) Neysla sauðfjárafurða á nýliðinni öld hefur verið áætluð út frá framboði annars vegar frá 1900-1940 (38) og hins vegar frá 1956-1995 (39). Þar kemur fram að neysla sauðfjárafurða, bæði kjöts og innmatar, var allnokkru meiri á fyrra skeiðinu en því síðara, sem var þó einnig veruleg og fór ekki að minnka að marki fyrr en 1986. Enda þótt rannsókn okkar á CJD hefjist ekki fyrr en 1960 er mikilsvert að hafa upplýsingar um neyslu frá fyrri hluta síðustu aldar því að sýnt hefur verið fram á að í príonsjúkdómnum Kuru sem smitast munnleiðis getur meðgöngutími sjúkdóms verið allt að hálf öld (40). Smitefnið er í mestu magni í heila riðuveiks fjár. Það kann að vera að fólk hafi lagt sér riðukindur til munns á öndverðri 20. öld þegar svarf að. En einkennalaust fé ber einnig smit. Lömb smitast að jafnaði við burð og bera það í ýmsum vefjum og blóði áður en það hefur borist í nægilegu magni til að valda riðueinkennum, en það tekur að jafnaði 2-4 ár. Það getur fundist í einhverju magni í heila áður en einkenni koma fram (41) og í eitilvef í eða tengdum meltingarvegi þar sem það finnst tiltölulega snemma (42). Það er athyglisvert að CJD fannst ekki í fólki búsettu á Mið-Norðurlandi, en þar hefur riða verið landlæg í ríflega öld og þéttleiki riðubæja verið mestur og þar tíðkaðist að neyta heila (43).
B) Bændur og búalið hafa auk þess að vera útsettir fyrir riðu vegna neyslu sauðfjárafurða verið útsettir fyrir riðusmiti við að handfjatla riðusýkt fé og við slátrun. Einnig við jarðvinnslu og heyskap því smitefnið getur haldist virkt í umhverfinu mjög lengi (44). Músatilraunir sýna að smitefni riðu getur borist frá rispum í húð til heila og valdið riðu (45) og því má ætla að smitefnið geti borist um sár á húð hjá mönnum.
Niðurstaða okkar er að riðusmit í sauðfé berist ekki í fólk og byggjum við það á lágri tíðni sCJD hérlendis þó riða hafi verið landlæg hér í nær 130 ár. Faraldsfræðileg rannsókn á riðu og CJD í Frakklandi sýndi heldur ekki fylgni milli neyslu sauðfjárafurða og CJD (46). Niðurstöður tveggja ítarlegra samanburðarrannsókna sýndu ekki fram á marktæk tengsl CJD við neyslu innmatar, þar með talinn heila (47), og neysla lambakjöts eða skepnuhirðingu (48). Tilgátan, sem var hvatinn að rannsókn okkar á CJD, það er að hin háa tíðni CJD í lýbískum Gyðingum í Ísrael væri vegna neyslu þeirra á augum, heila og mænu, reyndist ekki á rökum reist því að árið 1991 var sýnt fram á að um ættlægan sjúkdóm (gCJD) væri að ræða sem rekja mátti til stökkbreytingar í tákna 200 í príongeni (49). Orsök sCJD er eftir sem áður ókunn, en helstu tilgátur eru að rekja megi hana til stökkbreytingar í líkamsfrumu eða sjálfkrafa umbreytingar PrPC í PrPSc (12).
Þakkir
Við stöndum í þakkarskuld við marga, meðal annars Steinunni Árnadóttur lífeindafræðing fyrir aðstoð við vefjameinafræði, Birki Þór Birkisson sameindalíffræðing fyrir hjálp við vinnslu mynda, Thor Aspelund tölfræðing fyrir staðtölulega útreikninga, Hólmfríði Þorgeirsdóttur matvælafræðing og Guðmund Jónsson sagnfræðing fyrir upplýsingar um neysluvenjur, Matthew Bishop sameindalíffræðing í Edinborg fyrir athugun á tákna 129 og flokkun smitefnis. Verkefnið var styrkt af Evrópusambandsverkefnunum NEUROCJD (styrkur nr. QLK2 CT2001 02248) og PRIONET (styrkur nr. QLK2-CT-2000-00837). Rannsóknin var gerð með samþykki siðanefndar Landspítalans. Við tileinkum þessa grein minningu Gunnars heitins Guðmundssonar taugasjúkdómalæknis, en hann átti frumkvæðið að rannsókninni á CJD en lést áður en verkinu lauk.
Heimildir
1. Sánchez-Juan PJ. Etiologic and diagnostic facets of Creutzfeldt-Jakob disease. The effect of genes and environment. Chapter 2.1 The NEUROCJD" collaboration: CJD surveillance systems and results in 10 countries. (dissertation). Rotterdam, 2007.
2. Will RG, Alperovitch A, Poser S, et al. Descriptive epidemiology of Creutzfeldt-Jakob disease in six European countries, 1993-1995. Ann Neurol 1998; 43: 763- 7.
3. Ladogana A, Puopolo M, Croes EA, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia and Canada. Neurol 2005; 64: 1586-91.
4. Brown P, Gibbs CJ, Rodgers-Johnson P, et al. Human spongiform encephalopathy: The National Instiutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 1994; 35: 513-529.
5. Parchi P, Giese A, Capellari S, et al.Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999; 46: 224-33.
6. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216: 136-44.
7. Oesch B, Westaway D, Wälchli M, et al. A cellular gene encodes scrapie 27-30 protein. Cell 1985; 40: 735-46.
8. Büeler H, Fischer M, Lang Y, et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992; 356: 577-82.
9. Ma J, Wollmann R, Lindquist S. Neurotoxicity and neuro-degeneration when PrP accumulates in the cytosol. Science 2002; 298: 1781-5.
10. Roucou X, Guo Qi, Zhang Y, Goodyer CG, LeBlanc AC. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J Biol Chem 2003; 278: 40877-81.
11. Bragason BT, Pálsdóttir A. Interaction of PrP with NRAGE, a protein involved in neuronal apoptosis. Mol Cell Neurosci 2005; 29: 232-44.
12. Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95: 13363-83.
13. Weissmann C. The state of the prion. Nature Rev Microbiol 2004; 2: 861-71.
14. Büeler HR, Aguzzi A, Sailer A, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73: 1339-47.
15. Kirby L, Birkett CR., Rudyk H, Gilbert IH, Hope J. In vitro cell-free conversion of bacterial recombinant PrP to PrPres as a model for conversion. J.Gen.Virol. 2003; 84: 1013-1020.
16. Legname G, Baskakov IV, Nguyen H-OB, et al. Synthetic mammalian prions. Science 2004; 305: 673-6.
17. Castilla J, Saá P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell 2005; 121: 195-206.
18. Pálsson PA. Rida (Scrapie) in Iceland and its epidemiology. In: Prusiner SB and Hadlow WJ, eds Slow Transmissible Diseases of the Nervous System. Vol 1 Academic Press, New York, 1979: 357-66.
19. Hlíðar S. Riða Polyencephalitis. Í: Hlíðar S. Sauðfé og Sauðfjársjúkdómar. Akureyri, Þorsteinn M. Jónsson, 1937: 92-102.
20. Sigurðarson S. Scrapie eller rida i Island. Norsk Vet Tidskr 2000; 112: 408-13.
21. Kahana E, Alter M, Braham J, Sofer D. Creutzfeldt-Jakob disease: Focus among Libyan Jews in Israel. Science 1974; 183: 90-1.
22. Herzberg L, Herzberg BN, Gibbs Jr. CJ, Sullivan W, Amyx H, Gajdusek DC. Creutzfeldt-Jakob disease: Hypothesis for high incidence in Libyan Jews in Israel. Science 1974; 186: 848.
23. Alter M. Athugasemd við grein 22. Science 1974; 186: 848.
24. Collins S, Boyd A, Fletcher A, et al.Creutzfeldt-Jakob disease: diagnostic utility of 14-3-3 protein immunodetection in cerebrospinal fluid. J Clin Neurosci 2000; 7: 203-8.
25. Kovács GG, Gelpi E, Ströbel T, et al. Involvement of the endosomal-lysosomal system correlates with regional pathology in Creutzfeldt-Jakob disease. J Neuropath Exp Neurol 2007; 66: 628-36.
26. O´Rourke KI, Baszler TV, Miller JM, Spraker TR, Sadler-Riggleman I, Knowles DP. Monoclonal antibody F89/160.1.5. defines a conserved epitope on the ruminant prion protein. J Clin Microbiol 1998; 36: 1750-5.
27. Georgsson G, Gísladóttir E, Árnadóttir S. Quantitative assessment of the astrocytic response in natural scrapie of sheep. J Comp Path 1993; 108: 229-40.
28. Brownwell B, Oppenheimer DR. An ataxic form of subacute presenile polioencephalopathy (Creutzfeldt-Jakob disease). J Neurol Neurosurg Psychiatry 1965; 28: 350-61.
29. Cooper SA, Murray KL, Heath CA, Will RG, Knight RSG. Sporadic Creutzfeldt- Jakob disease with cerebellar ataxia at onset in the UK. J Neurol Neurosurg Psychiatry 2006; 77: 1273-5.
30. Laplanche J-L, Delasnerie-Laupretre N, Brandel JP, et al. Molecular genetics of prion diseases in France. Neurol 1994; 44: 2347-51.
31. Schulz-Schaeffer WJ, Giese A, Windl O, Kretzschmar HA. Polymorphism at codon 129 of the prion proetein gene determines cerebellar pathology in Creutzfeldt-Jakob disease. Clin Neuropathol 1996; 15: 353-7.
32. Henry C, Lowman A, Will RG. Creutzfeldt- Jakob disease in elderly people. Age Ageing 2002; 31: 7-10.
33. Murray K, Ritchie DL, Bruce M, et al.Sporadic Creutzfeldt-Jakob disease in two adolescents. J Neurol Neurosurg Psychiatry 2008; 79: 14-8.
34. Budka H, Aguzzi A, Brown P, et al. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol 1995; 5: 459-66.
35. Georgsson G, Tryggvason T, Jónasdóttir AD, Guðmundsson S, Þorgeirsdóttir S. Polymorphism of PRNP codons in the normal Icelandic population. Acta Neurol Scand 2006; 113: 419-25.
36. Shibuya S, Higuchi J, Shin R-W, Tateishi J, Kitamoto T. Codon 219 lys allele of PRNP is not found in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1998; 43: 826-8.
37. Aguzzi A, Heikenwalder M, Miele G. Progress and problems in the biology, diagnostics, and therapeutics of prion diseases. J Clin Invest 2004; 114: 153-160.
38. Jónsson G. Changes in food consumption in Iceland, 1770-1940. Scand EconomHist Rev 1998; 46: 24-41.
39. Þorgeirsdóttir H. Per Capita Supply of Food in Iceland 1956-1995. Master thesis (1999) Human Nutrition Department of Food Science, Faculty of Science, University of Iceland.
40. Collinge J, Whitfield J, McKintosh E, et al. Kuru in the 21st century-an acquired human prion disease with very long incubation periods. Lancet 2006; 367: 2068-74.
41. Þorgeirsdóttir S, Georgsson G, Reynisson E, Sigurðarson S, Pálsdóttir A.Search for healthy carriers of scrapie: an assessment of subclinical infection of sheep in an Icelandic scrapie flock by three diagnostic methods and correlation with PrP genotypes. Arch Virol 2002; 147: 709-22.
42. Georgsson G, Adolfsdóttir JA, Pálsdóttir A, Jörundsson E, Sigurðarson S, Þorgeirsdóttir S. High incidence of subclinical infection of lymphoid tissues in Scrapie affected sheep flocks. Arch Virol 2008; 153: 637-44.
43. Gísladóttir H. Íslensk matarhefð. Mál og menning, Reykjavík 1999.
44. Georgsson G, Sigurðarson S, Brown P. Infectious agent of sheep scrapie may persist in the environment for at least 16 years. J Gen Virol 2006; 87: 3737-40.
45. Mohan J, Brown KL, Farquhar CF, Bruce ME, Mabbott NA. Scrapie transmission following exposure through the skin is dependent on follicular dendritic cells in lymphoid tissues. J Dermatol Sci 2004; 35: 101-11.
46. Chatelain J, Cathala F, Brown P, Raharison S, Court L, Gajdusek DC. Epidemiologic comparisons between Creutzfeldt-Jakob disease and scrapie in France during the 12-year period 1968-1979. J Neurol Sci 1981; 51: 329-37.
47. Wientjens DPWM, Davanipour Z, Hofman A, et al. Risk factors for Creutzfeldt-Jakob disease: A reanalysis of case-control studies. Neurol 1996; 46: 1287-91.
48. Van Duijn CM, Delasnerie-Laupretre N, Masullo C, et al. Case-control study of risk factors of Creutzfeldt-Jakob Disease in Europe during 1993-95. Lancet; 352: 1081-5.
49. Korczyn AD, Chapman J, Goldfarb LG, Brown P, Gajdusek DC. A mutation in the prion protein gene in Creutzfeldt-Jakob disease in Jewish patients of Libyan, Greek, and Tunisian origin. Ann NY Acad Sci 1991; 640: 171-6. 1) Kvennasviði, 2) myndgreiningarsviði Landspítala.