
10. tbl. 104. árg. 2018
Fræðigrein
Peutz-Jeghers-heilkenni með garnasmokkun
Ágrip
Hér er lýst tilfelli sjúklings með staðfest Peutz-Jeghers-heilkenni á grundvelli jákvæðrar vefjagreiningar samhliða litabreytingum á vörum. Þessi sjúklingur greindist ekki fyrr en hann fékk innsmokkun á mjógirni. Hægt hefði verið að greina hann fyrr á ævinni vegna fyrrgreindra litabreytinga og sögu um tíð kviðverkjaköst.
Barst til blaðsins 16. apríl 2018, samþykkt til birtingar 3. júlí 2018.
Höfundar fengu samþykki sjúklings fyrir þessari umfjöllun og birtingu.
Inngangur
Peutz-Jeghers-heilkenni er sjaldgæfur ríkjandi A-litninga erfðasjúkdómur sem lýsir sér í sepamyndun í meltingarvegi auk litabreytinga á húð og slímhúð. Sjúklingar með heilkennið eru í aukinni hættu á að fá krabbamein á lífsleiðinni, auk annarra kvilla í tengslum við sepamyndun.1,2
Tilfelli
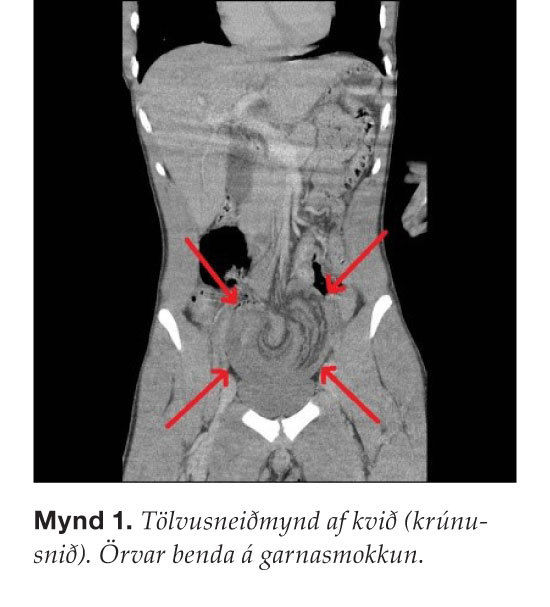
21 árs gamall hraustur karlmaður sem hafði nokkrum sinnum fengið væg kviðverkjaköst leitaði á bráðamóttöku vegna skyndilegra, slæmra, dreifðra kviðverkja, ógleði og uppkasta sem höfðu varað í tvær klukkustundir. Hann var illa haldinn af verkjum en kviðurinn var þó mjúkur og án þreifieymsla við fyrstu skoðun og garnahljóð voru eðlileg. Verkirnir versnuðu hratt, maðurinn var kaldsveittur og fölur en ítrekuð kviðskoðun var eðlileg. Hækkun á hvítum blóðkornunum var væg (13,5 þúsund) en C-reaktíft prótein var eðlilegt. Fengin var tölvusneiðmynd af kvið sem sýndi garnasmokkun á 25-30 cm bili neðarlega í kvið ( mynd 1 ).
{kind=link}
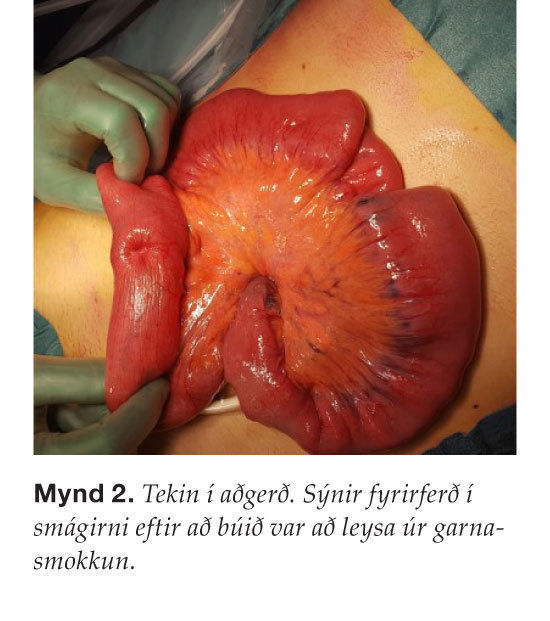
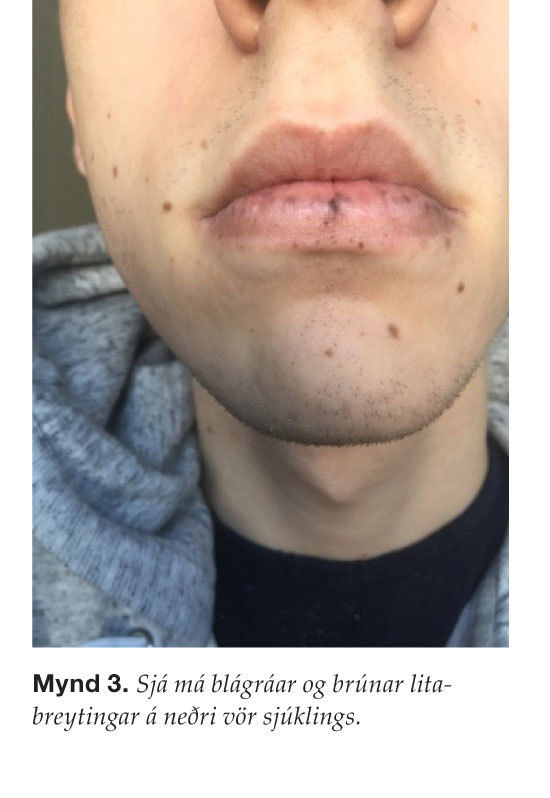
Gerð var kviðarholsaðgerð þar sem garnasmokkun sást á um það bil hálfum metra af smágirni. Leyst var úr garnasmokkuninni og fyrirferð í smágirninu þreifaðist og var vendi-punktur garnasmokkunarinnar ( mynd 2 ). Gert var hlutabrottnám á smágirni og fyrirferðin fjarlægð. Vefjagreining sýndi sepa með einkennandi útlit fyrir sepamyndun í Peutz- Jeghers-heilkenni og engin merki um æxlisvöxt í aðlægum eitlum. Þegar sjúklingur var skoðaður með tilliti til þessa sjúkdóms sáust vel litabreytingar á vörum sem fjölskylda sjúklingsins hafði tekið eftir hjá honum í æsku ( mynd 3 ). Auk þess voru tvær litabreytingar til staðar í slímhúð munnhols en engar litabreytingar fundust á iljum eða lófum. Rannsókn hjá erfðaráðgjöf staðfesti að um Peutz-Jeghers-heilkenni væri að ræða, líklegast sporadísk erfðabreyting þar sem ættarsaga var engin. Gerð var segulómmynd af kviði til eftirlits sem sýndi ekki fram á æxlisvöxt. Fyrirhugað er krabbameinseftirlit með maga- og ristilspeglun á eins til tveggja ára fresti auk árlegrar skimunar með segulómun af eistum og á eins til tveggja ára fresti af brisi.
{kind=link}
{kind=link}
Umræða
Peutz-Jeghers-heilkenni var fyrst lýst árið 1921 af Jan L. Peutz- og svo að nýju árið 1944 af Harold J. Jeghers. Heilkennið er sjaldgæfur ríkjandi A-litninga erfðasjúkdómur með háa tíðni svipgerðar sem lýsir sér í sepamyndun í meltingarvegi auk litabreytinga á húð og slímhúð.1-3 Fjölskyldusaga er til staðar í um 55% tilfella auk þess sem tíðni krabbameins í fyrstu gráðu ættingjum er hærri en í almennu þýði og er þess vegna mælt með að þessir einstaklingar gangist undir krabbameinseftirlit og nánir ættingjar þeirra fái erfðaráðgjöf með tilheyrandi genaprófun.4-6
Algengi Peutz-Jeghers-heilkennis er 1:8300 til 1:200.0007 og er enginn munur milli kynja. Heilkennið er einkennalaust í 50% tilfella við greiningu. Algengustu fylgikvillar sjúkdómsins eru: garnasmokkun, blæðing frá sepum með fylgjandi blóðskorti og stífla í meltingarvegi vegna sepa. Meðalaldur fyrstu einkenna frá meltingarfærum er 13 ár. Allt að 69% einstaklinga með Peutz-Jeghers-heilkenni fá garnasmokkun á lífsleiðinni og þá oftast í dausgörn. Í um það bil 90% tilfella lýsa sjúklingar þá miklum kviðverk en í 40% tilfella eru þeir með einkenni smágirnisstíflu. Einnig geta þeir verið með blóð í hægðum, niðurgang og blóðskort.3,8-11
Sepamyndunin er í formi vaxtarvillusepa (hamartomatous polyps) og getur komið fram hvar sem er í meltingarveginum en er algengust í smágirni.8,12 Separnir geta einnig myndast utan meltingarvegar, til dæmis í þvagblöðru, nýrnaskjóðum, lungum og nefkoki. Sjúklingar með Peutz-Jeghers-heilkenni eru í aukinni hættu að mynda krabbamein sökum sepamyndunar en 38-66% þeirra greinast með krabbamein í meltingarvegi á ævinni.1
Yfir 95% sjúklinga með Peutz-Jeghers-heilkenni eru með fyrrnefndar litabreytingar. Þær koma oftast fram á unga aldri og eru jafnvel til staðar við fæðingu. Þá eru þær helst á vörum, við munn og í munnslímhúð en einnig í lófa, undir iljum, við nef, endaþarm og kynfæri. Um er að ræða blá-gráa eða brúna fleti, 1-5 mm að stærð, sem koma fram snemma á æviskeiði og eru ekki taldir vera forstig krabbameins.8,13 Sjúklingurinn í okkar tilfelli reyndist vera með litabreytingar í kringum munn, á vörum og einnig í munnslímhúð, en ekki á iljum eða lófum.
Sjúklingur skal uppfylla eitt af eftirfarandi skilgreiningaratriðum til að uppfylla klínísk greiningarskilmerki fyrir Peutz-Jeghers-heilkenni:14,15
- Tveir eða fleiri separ sem samræmast Peutz-Jeghers-heilkenni á vefjagreiningu.
- Fjölskyldusaga um Peutz-Jeghers-heilkenni auk sepa hjá sjúklingi sem samræmist heilkenninu á vefjagreiningu.
- Fjölskyldusaga um Peutz-Jeghers-heilkenni með litabreytingum hjá sjúklingi sem samræmast heilkenninu.
- Einkennandi litabreyting ásamt sepa sem samræmist Peutz- Jeghers-heilkenni á vefjagreiningu.
Sjúklingur í okkar tilfelli var með einkennandi litabreytingar ásamt sepa sem samræmdist Peutz-Jeghers-heilkenni á vefjagreiningu og uppfyllti því skilmerki Alþjóðaheilbrigðisstofnunarinnar (WHO) fyrir sjúkdóminn.
Einstaklingar sem greinast með heilkennið eiga að gangast undir krabbameinseftirlit sem felur í sér eftirfarandi:6
- Maga- og ristilspeglun auk speglunar, hylkisrannsóknar eða segulómunar á smágirni frá 8 ára aldri. Þessar rannsóknir skal framkvæma á þriggja ára fresti ef separ eru til staðar. Annars skal endurtaka rannsókn við 18 ára aldur.
- Klínísk brjóstaskoðun á 6 mánaða fresti frá 18 ára aldri auk brjóstamyndatöku árlega frá 25 ára aldri.
- Skoðun á kvenlíffærum með ómun, grindarbotnsskoðun með stroki og CA-125 mælingu í blóði árlega frá 18-20 ára aldri.
- Segulómskoðun, ómun í speglun eða segulómun af gall- og brisgangi á eins til tveggja ára fresti frá 30 ára aldri.
- Klínísk eistnaskoðun, auk ómunar ef klínísk skoðun er afbrigðileg, árlega frá fæðingu eða unglingsárum.
Niðurstaða
Niðurstaðan sem draga má af þessu tilfelli er að ávallt skal hafa Peutz-Jeghers-heilkenni í huga ef einstaklingur hefur fengið endurtekna kviðverki frá barnæsku með fyrrnefndum litabreytingum. Sér í lagi ef hann hefur fengið blæðingu um endaþarm eða reynist vera með blóðskort. Það væri hagur í því að ná þessum sjúklingum í viðeigandi eftirlit áður en þeir fá smágirnisstíflu eða garnasmokkun.
Heimildir
| 1. van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol 2010; 105: 1258-64; author reply 65. https://doi.org/10.1038/ajg.2009.725 PMid:20051941 |
|
| 2. Georgescu EF, Stänescu L, Simionescu C, Georgescu I, Ionescu R, Florescu G. Peutz-Jeghers syndrome: case report and literature review. Rom J Morphol Embryol 2008; 49: 241-5. PMid:18516333 |
|
| 3. Mozaffar M, Sobhiyeh MR, Hasani M, Fallah M. Peutz-Jeghers syndrome without mucocutaneous pigmentation: a case report. Gastroenterol Hepatol Bed Bench 2012; 5: 169-73. PMid:24834220 PMCid:PMC4017480 |
|
| 4. Sökmen HM, Ince AT, Bölükbas C, Kilic G, Dalay R, Kurdas OO. A Peutz-Jeghers syndrome case with iron deficiency anemia and jejuno-jejunal invagination. Turk J Gastroenterol 2003; 14: 78-82. PMid:14593545 |
|
| 5. Schulmann K, Pox C, Tannapfel A, Schmiegel W. The patient with multiple intestinal polyps. Best Pract Res Clini Gastroenterol 2007; 21: 409-26. https://doi.org/10.1016/j.bpg.2006.11.003 PMid:17544108 |
|
| 6. McGarrity TJ AC, Baker MJ. Peutz-Jeghers Syndrome. Seattle (WA): University of Washington, Seattle, 1993-2018. | |
| 7. Lindor NM, Greene MH. The concise handbook of family cancer syndromes. Mayo Familial Cancer Program. J Natl Cancer Inst 1998; 90: 1039-71. https://doi.org/10.1093/jnci/90.14.1039 PMid:9672254 |
|
| 8. Utsunomiya J, Gocho H, Miyanaga T, Hamaguchi E, Kashimure A. Peutz-Jeghers syndrome: its natural course and management. Johns Hopkins Med J 1975; 136: 71-82. PMid:1117595 |
|
| 9. Hinds R, Philp C, Hyer W, Fell JM. Complications of childhood Peutz-Jeghers syndrome: implications for pediatric screening. J Pediatr Gastroenterol Nutr 2004; 39: 219-20. https://doi.org/10.1097/00005176-200408000-00027 PMid:15269641 |
|
| 10. van Lier MG, Mathus-Vliegen EM, Wagner A, van Leerdam ME, Kuipers EJ. High cumulative risk of intussusception in patients with Peutz-Jeghers syndrome: time to update surveillance guidelines?Am J Gastroenterol 2011; 106: 940-5. https://doi.org/10.1038/ajg.2010.473 PMid:21157440 |
|
| 11. Pétursdóttir K, Rósmundsson Þ, Hannesson PH, Möller PH. Garnasmokkun hjá börnum á Íslandi. Læknablaðið 2013; 99: 77-81. PMid:23486679 |
|
| 12. Jeghers H, Mc KV, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med 1949; 241: 1031-6. https://doi.org/10.1056/NEJM194912222412501 https://doi.org/10.1056/NEJM194912292412601 PMid:15398245 |
|
| 13. Gammon A, Jasperson K, Kohlmann W, Burt RW. Hamartomatous polyposis syndromes. Best Pract Res Clin Gastroenterol 2009; 23: 219-31. https://doi.org/10.1016/j.bpg.2009.02.007 PMid:19414148 PMCid:PMC2678968 |
|
| 14. Aaltonen LA. Hereditary intestinal cancer. Semin Cancer Biol 2000; 10: 289-98. https://doi.org/10.1006/scbi.2000.0148 PMid:10966851 |
|
| 15. Aretz S, Stienen D, Uhlhaas S, Loff S, Back W, Pagenstecher C, et al. High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat 2005; 26: 513-9. https://doi.org/10.1002/humu.20253 PMid:16287113 |
|
| 16. Latchford AR, Neale K, Phillips RK, Clark SK. Juvenile polyposis syndrome: a study of genotype, phenotype, and long-term outcome. Dis Colon Rectum 2012; 55: 1038-43. https://doi.org/10.1097/DCR.0b013e31826278b3 PMid:22965402 |
|
| 17. Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW, et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 2015; 110: 223-62; quiz 63. https://doi.org/10.1038/ajg.2014.435 PMid:25645574 PMCid:PMC4695986 |
|