
12. tbl. 94. árg. 2008
Fræðigrein
Slímseigjusjúkdómur (cystic fibrosis): meingerð, greining og meðferð - Yfirlitsgrein
Ágrip
Slímseigjusjúkdómur er arfgengur víkjandi sjúkdómur sem orsakast af stökkbreytingu í CFTR geninu, en próteinafurð þess myndar jónagöng sem stýra salt- og vökvabúskap í þekjufrumum. Yfir 1500 stökkbreytingar eru þekktar. Tíðni sjúkdómsins er 1/2.000-3.000 í evrópskum þjóðum. Galli í CFTR hefur áhrif á seyti þekjuvefs og upptöku um hann í ýmsum líffærum. Áhrifin eru mismunandi eftir því hvaða líffæri á í hlut, en teppa í seyti útkirtla er sameiginlegt vandamál. Helstu líffæri sem verða fyrir áhrifum eru öndunarfæri, bris, meltingarfæri og svitakirtlar. Sjúkdómurinn greinist oftast á fyrstu mánuðum lífs og eru vanþrif, saltur sviti og hægðabreytingar vegna vanmeltingar á fitu algeng birtingarmynd. Til að greina sjúkdóminn er mögulegt að gera svitapróf, erfðaefnispróf og fleira. Öndunarfærasjúkdómur er oftast alvarlegasti hluti sjúkdómsins, með langvinnum sýkingum með ýktu bólgusvari auk þess sem lungnaslím er þykkara og öðruvísi samsett en í heilbrigðum. Samspil sýkla við þekjuvef veldur því að S.aureus og síðar P.aeuruginosa breytast í slímmyndandi form sem erfitt er að uppræta og gerir þessar tegundir því helstu skaðvaldana. Helstu lyf við öndunarfæraeinkennum eru sýklalyf, berkjuvíkkandi lyf, lyf sem auka seyti loftvega og bólgueyðandi lyf. 90% sjúklinga hafa vanstarfsemi briss sem er meðhöndlað með brisensímum. Gott næringarástand er mikilvæg forsenda allrar meðferðar. Horfur sjúklinga hafa batnað mikið undanfarna áratugi með vaxandi þekkingu og er meðalaldur nú 37 ár í Bandaríkjunum.
Inngangur
Slímseigjusjúkdómur (cystic fibrosis) er arfgengur sjúkdómur sem hefur áhrif á mörg líffæri, einkum öndunarfæri, bris og meltingarfæri. Einnig hafa sjúklingar óvenju saltan svita. Sjúkdómnum var fyrst lýst á miðöldum en þá var hinn salti sviti bendlaður við galdra og börn með sjúkdóminn stundum talin andsetin. Síðan þá hefur þekkingu um sjúkdóminn fleygt fram. Á fimmta áratug síðustu aldar kom í ljós að sjúkdómurinn er erfðasjúkdómur sem erfist víkjandi. Árið 1983 var sannað að sjúkdómnum veldur brenglun á flutningi klóríðjóna yfir frumuhimnur1 en 1989 var sjúkdómsgenið og próteinafurð þess skilgreint.2
Orsök sjúkdómsins er stökkbreyting í CFTR (cystic fibrosis transmembrane conductance regulator) geni á litningi 7, en CFTR er prótein sem myndar klóríðgöng í himnum þekjuvefsfrumna. Fyrsta stökkbreytingin og sú algengasta fannst árið 1989 (ΔF508) en síðan þá hafa að minnsta kosti 1500 stökkbreytingar fundist. Erfðirnar eru víkjandi (autosomal recessive).2, 3 Tíðni sjúkdómsins er 1/2000-3000 í þjóðum af evrópskum uppruna4, en íslenskar rannsóknir benda til þess að tíðnin sé mun lægri hérlendis.5
Meinmyndun
Ýmsar kenningar eru um það hvernig stökkbreyting í CFTR veldur sjúkdómseinkennum og meinafræðilegum breytingum hjá sjúklingum með slímseigjusjúkdóm. CFTR próteinið er staðsett á efra borði þekjuvefsfrumna í húð, öndunarfærum, meltingarfærum og þvag- og kynfærum, og er jónagöng fyrir klóríðjónir. Galli í því hefur þannig áhrif á seyti þekjuvefsins og upptöku um hann.6,7 Áhrifin eru mismunandi eftir því hvaða líffæri á í hlut og einnig er þekkt að mismunandi stökkbreytingar hafa mismunandi áhrif á líffærakerfin.1
Til dæmis eru sjúklingar arfhreinir fyrir ?F508 stökkbreytingunni ávallt með vanstarfsemi briss en brisstarfsemi helst nær óskert hjá sjúklingum með sumar aðrar stökkbreytingar. Mismunurinn stafar af því hvar í myndunarferli próteinsins gallinn er.8, 9 Þó er einnig þekkt að sjúklingar með sömu arfgerð geta haft gjörólíka klíníska mynd, og talið er að umhverfisþættir og önnur gen og afurðir þeirra hafi þar áhrif.6, 9
CFTR próteinið, sem tilheyrir flokki próteina er nefnast ATP-binding cassette, er virkjað af cAMP (cyclic AMP).1,7 Talið er að CFTR hafi áhrif á önnur jónagöng í frumuhimnunni annaðhvort beint eða óbeint með því að hafa áhrif á himnuspennu. Ein afleiðing þessa er sú að í öndunarfæraþekju sjúklinga með slímseigjusjúkdóm er tekið upp meira af natríumjónum í gegnum ENaC (epithelial sodium channel).6 Seyti svitakirtla er saltara í sjúklingum með slímseigjusjúkdóm, og þetta er notað til að greina sjúkdóminn með sérstöku svitaprófi (10). Jafnvægi fleiri jóna yfir frumuhimnuna er einnig talið verða fyrir áhrifum af göllum í CFTR. Þetta ójafnvægi í jónaflutningi er talið eiga mestan þátt í meingerð sjúkdómsins og veldur teppu í seyti útkirtla (exocrine glands).6, 7
Upphafseinkenni og greining
Algengt er að vanþrif hjá ungbörnum séu fyrsta vísbending um slímseigjusjúkdóm. Sjaldnar koma fram öndunarfæraeinkenni eða bjúgur vegna þess að upptaka próteina um meltingarveg er minni en eðlilegt er. Hægðir eru oftast fitugar og illa lyktandi vegna vanmeltingar. Hjá nýburum með barnabiks-garnalömun (meconium ileus) ætti alltaf að útiloka slímseigjusjúkdóm, en garnalömun getur myndast vegna þykkara barnabiks en í heilbrigðum börnum.6, 10 Í töflu I eru rakin helstu einkenni sjúkdómsins.

Slímseigjusjúkdómur greinist oftast á fyrstu mánuðum lífs. Sums staðar í heiminum fer fram skimun fyrir sjúkdómnum hjá nýburum með því að mæla próteinið trypsin í blóði (immune reactive trypsin) sem er til staðar í auknu magni hjá börnum með slímseigjusjúkdóm, en það er ekki gert hérlendis.6, 11 Svokallað svitapróf er gert ef grunur leikur á að um sjúkdóminn sé að ræða en þá er sviti framkallaður á húð, og styrkur natríum- og klóríðjóna mældur í svitanum. Erfðaefnispróf eru síðan gerð til að staðfesta sjúkdóminn og greina hvaða stökkbreytingar í CFTR geninu eru til staðar.10 Klassísk greiningarskilmerki eru eftirfarandi:
1. Klínísk: Dæmigerð klínísk einkenni eða fjölskyldusaga.
2. Rannsóknir: Svitapróf, erfðaefnispróf eða rafhrif í nefslímhúð (nasal voltage).
Einhver af skilmerkjunum í 1. og 2. þurfa að vera til staðar til þess að greina sjúkdóminn. Aðrar mælingar sem gefa vísbendingar um sjúkdóminn eru til dæmis greining á sæði, mæling á ensímum í saur, mæling á próteinum í berkjuskolvökva en þær eru ekki notaðar að staðaldri.10
Sumir sjúklingar uppfylla ekki öll greiningarskilmerki og geta til dæmis verið með klassísk einkenni og stökkbreytingar í CFTR en eðlilegt svitapróf. Aðrir eru ekki með dæmigerð einkenni heldur vægari sjúkdómsmynd með brisbólgu, galla í sáðrás og sepum í nefi. Hjá slíkum sjúklingum ætti að gera erfðaefnisrannsókn til þess að erfðaráðgjöf sé möguleg.10 Að lokum má nefna að lítill hópur er með klínísk einkenni slímseigjusjúkdóms, þar með talið jákvætt svitapróf, en enga stökkbreytingu í CFTR. Ástæða sjúkdómsins í þessum sjúklingum er ókunn, en líklega er um að ræða óþekkta stökkbreytingu.12 Eins og búast má við eru þeir sem greinast eftir táningsár yfirleitt með vægari sjúkdóm en þeir sem greinast ungir.13 Hins vegar er mjög mikilvægt að greina börn með sjúkdóminn sem fyrst og rannsóknir hafa sýnt að snemmgreining tengist betri horfum.14
Meinmyndun, einkenni og meðferð
Öndunarfæri
Þrátt fyrir að stökkbreyting í CFTR valdi oftast mestum einkennum frá öndunarfærum þarf meðferð sjúkdómsins að vera heildræn og taka á öllu sem hægt er að meðhöndla miðað við einkenni sjúklinga og þá meðferðarmöguleika sem bjóðast. Mjög mikilvægt er einnig að sjúklingar nærist vel, og hefur vannæring áhrif á allar hliðar sjúkdómsins.6, 15
Helstu öndunarfæraeinkenni stafa af langvinnum bakteríu- og veirusýkingum með bólgu-svari þar sem daufkyrningar (neutrophiles) eru mest áberandi. Þessu fylgir langvinnur hósti með uppgangi og mæði.6, 7 Það jóna-ójafnvægi sem myndast vegna stökkbreytingar í CFTR veldur þykkara slími í berkjum og breyttu sýrustigi sem bætir vaxtarskilyrði öndunarfærasýkla og veikir getu daufkyrninga til þess að drepa þá. Lungun eiga einnig erfiðara með að losa sig við þykka slímið sem myndast.6, 7, 16 Langtímaafleiðing þessa er versnandi geta öndunarfæra til að sinna hlutverki sínu, aukin teppa, versnandi loftskipti og að lokum öndunarbilun.17 Þessar breytingar valda því að flestir sjúklingar fá einhvern tíma berkjuauðreitni, líkt og í astma.15 Með tímanum myndast skemmdir í berkjuveggjum, berkjur víkka og berkjuskúlk (bronchiectasis) kemur fram á lungnamynd. Langvinnar kinnholubólgur og separ í nefi eru algengir fylgikvillar, en sjúklingar geta einnig fengið loftbrjóst eða blóðhósta.6
Ákveðnar bakteríur eru einkennandi fyrir lungnasýkingarnar. Í byrjun eru það oftast Staphylococcus aureus og Haemophilus influenzae, en seinna í sjúkdómsganginum er Pseudomonas aeruginosa helsti skaðvaldurinn og rannsóknir hafa sýnt að meira en 70% af fullorðnum sjúklingum eru með sýkilinn í loftvegum að staðaldri.18 Aðrar bakteríur sem ræktast oftar úr lungum slímseigjusjúklinga en heilbrigðra einstaklinga eru Stenotrophomonas maltophilia, Mycobacteria aðrar en M.tuberculosis, Alcaligenes xylosoxidans og Burkholderia cepacia complex, en sýklun með þeirri síðastnefndu er tengd hraðri versnun sjúkdómsins.6, 17, 19 P. aeruginosa og S. aureus eru ekki slímmyndandi (non-mucoid) þegar sýklarnir berast í lungun, en vaxtarskilyrði þar eru hvati að breytingu þessara bakteríutegunda yfir í slímmyndandi (mucoid) svipgerð.6, 20 Þessi svipgerð er lítt næm fyrir sýklalyfjum og þess vegna er afar erfitt að uppræta hana. Slímmyndandi bakteríur lifa þannig í sambýli (biofilm) með hýslinum með tilheyrandi langvinnu bólgusvari og skaðlegri losun á ensímum og bólgumiðlum, en valda hins vegar sjaldan ífarandi sýkingu með blóðsýkingu (sepsis).20
Reglulegt eftirlit er lykilatriði í meðferð sjúklinga með slímseigjusjúkdóm. Fylgjast þarf vel með einkennum sjúklinga og er það gert með reglulegum læknisheimsóknum (sex sinnum á ári hér á landi), líkamsskoðun, ræktunum á hráka og lungnastarfsemisprófum. Mælt er með ræktun á hráka að minnsta kosti fjórum sinnum á ári. Þegar lungnasjúkdómur er langt genginn getur verið nauðsynlegt að mæla súrefnismettun, bæði með mettunarmæli útlægt og jafnvel með blóðgasmælingu þegar það á við. Þessar mælingar gefa til kynna hvort þörf er á meðferð með súrefni. Röntgenmynd af lungum er tekin ef um bráða versnun á einkennum er að ræða og grunur um lungnabólgu, loftbrjóst eða samfall á lunga, en annars árlega. Lungnastarfsemispróf eru einnig mikilvæg í að meta versnanir, svo og öndunaræfingar sem hjálpa til við hreinsun slíms úr lungum.15, 17 Tölvusneiðmyndir gegna hins vegar lykilhlutverki í greiningu berkjuskúlks.21
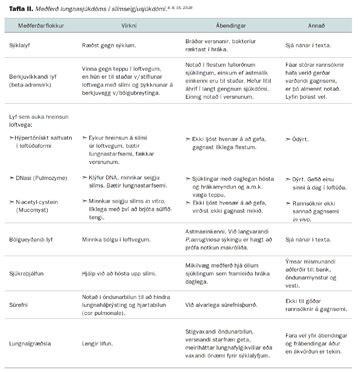
Meðferð við lungnasjúkdómi hjá sjúklingum með slímseigjusjúkdóm má gróflega skipta í eftirfarandi flokka:
- Sýklalyf
- Berkjuvíkkandi lyf
- Lyf sem auka hreinsun loftvega
- Bólgueyðandi lyf
- Sjúkraþjálfun
- Súrefni
- Lungnaígræðsla
Auk þess skipta athafnir og dagleg umgengni miklu máli. Dæmi um þetta er sú regla að sjúklingar sem ekki hafa langvinna P.aeruginosa sýkingu mega alls ekki umgangast þá sem hafa slíka sýkingu.15 Einnig er talið mikilvægt fyrir sjúklinga að stunda almenna hreyfingu eins oft og auðið er og talið að slímið í loftvegunum losni frekar við áreynslu. Vaxandi áhersla er lögð á meðferð í heimahúsi.22
Hér verður farið yfir sýklalyfjameðferð og einnig rætt stuttlega um meðferð með bólgueyðandi lyfjum og hýpertónísku saltvatni. Yfirlit yfir aðra flokka meðferðar má sjá í töflu II.
Sýklalyf
Bráð versnun á hósta, hrákamyndun eða mæði, er algengasta ástæða þess að sjúklingar leita læknis utan reglulegra heimsókna. Við þessar aðstæður getur öndunarmæling (FEV1) einnig lækkað, auk versnunar á einkennum. Meðferð skal þá hefja með sýklalyfjum og berkjuvíkkandi lyfjum, en sýklalyf eru einnig gefin ef sýkill ræktast úr hráka án þess að einkenni séu til staðar. Meðferð skal standa yfir í 2-4 vikur.6, 15, 17
Best er að bíða eftir sýkla- og næmisrannsókn þar til meðferð með sýklalyfi er hafin. Ef veikindi eru mjög bráð, skal gefa tvö sýklalyf sem bæði vinna á P.aeruginosa, til dæmis flúor-ókínólón og amínóglýkósíð. Þessa lyfjablöndu skal einnig nota ef alvarleg P.aeruginosa sýking er staðfest með sýklaræktun, en í vægari tilfellum má einungis nota annað lyfið. Við S. aureus sýkingu skal nota viðeigandi sýklalyf, til dæmis díkloxacillín.15, 17 Hafa skal í huga að oft þarf stærri skammta og fleiri gjafir sýklalyfja hjá sjúklingum með slímseigjusjúkdóm. Orsök þessa er meðal annars sú að dreifingarrúmmál vatnssækinna lyfja, þar með talið flestra sýklalyfja, er aukið þar sem sjúklingarnir eru oft vannærðir og með hlutfallslega lítinn fituvef.27, 28 Venjan er að gefa ekki sama sýklalyfið við tvær versnanir í röð vegna hættu á myndun ónæmis. Byrjað er á meðferð í töfluformi, en stundum getur þurft að leggja sjúklinga inn til meðferðar með sýklalyfi í æð ef versnunin er alvarleg, svarar ekki sýklalyfjum í töfluformi eða ef næmispróf sýnir ónæmi fyrir sýklalyfjum sem unnt er að gefa í töfluformi.15
Í sumum tilvikum eru sýklalyf í töflum gefin stöðugt þar sem sýklalyfjakúrar við versnanir duga ekki til þess að uppræta lungnasýkingar. Þetta er umdeild meðferð vegna hættu á myndunar ónæmis fyrir sýklalyfjum og gagnsemi hefur ekki verið sönnuð.29 Rannsóknir á tobramycin-úða sýna hins vegar að hann gagnast sjúklingum með meðalslæman sjúkdóm og króníska P. aeruginosa sýkingu, með því að bæta lungnastarfsemi, minnka þéttni P. aeruginosa í hráka og fækka innlagnardögum.30
Bólgueyðandi lyf
Bólgan í berkjum sjúklinga með slímseigju-sjúkdóm er svar við árás sýkla og í þeim skilningi nauðsynleg, en á hinn bóginn benda rannsóknir til þess að ýkt bólgusvar og ójafnvægi í bólgumiðlum valdi hluta af einkennum sjúkdómsins, eins og hefur verið rætt um hér að framan.4, 31 Til þess að draga úr bólgu hafa verið reynd nokkur lyf, meðal annars barksterar, makrólíðar og íbúprófen.
Barksterar í töfluformi gagnast við versnunum en aukaverkanir, svo sem hækkun blóðsykurs, dreri (cataract) og vaxtarskerðing, hindra almenna notkun. Þeir eru hins vegar notaðir í vissum tilvikum, til dæmis ef astmaeinkenni eru mikil, hjá sjúklingum með „allergic bronchopulmonary aspergillosis“ og í sumum tilfellum hjá bráðveikum sjúklingum.32, 33 Barksterar í innúða eru notaðir ef astmaeinkenni eru til staðar, en rannsaka þarf betur hvort þeir eru gagnlegir við aðrar aðstæður.34
Í ljós hefur komið að notkun makrólíða við öndunarfærasýkingum í slímseigjusjúkdómi bætir lungnastarfsemi og horfur, óháð því hvort sýkingin er upprætt. Leiddar hafa verið líkur að því að makrólíðar hafi bólgueyðandi áhrif, en erfitt hefur reynst að sanna það með klínískum rannsóknum. Rannsóknir sýna einnig að makrólíðinn azithromycin breytir starfsemi og tjáningu þétttengslapróteina í þekjuvef lungna in vitro.35 Þessi prótein stjórna flæði jóna og vökva gegnum þekjuna og er því hugsanlegt að jákvæð áhrif makrólíða á lungnasjúkdóm slímseigju stafi af nokkurs konar leiðréttingu á jónaflutningi sem gæti bætt fyrir vanstarfsemi CFTR.35 Ítarlegar rannsóknir þarf til að skýra þetta samband betur. Mælt er með að íhuga notkun makrólíða til lengri tíma hjá sjúklingum eldri en 6 ára sem eru með króníska P. aeruginosa sýkingu og svara ekki annarri meðferð.15, 36
Hýpertónískt saltvatn
Gallinn í CFTR próteininu er talinn valda lækkun á styrk NaCl í berkjuvökva. Þetta leiðir til vatnsskorts sem veldur því að berkjuslím þykknar og verður ákjósanleg bólfesta sýkla. Eftir sýkingu þykknar slímið enn meira og vítahringur myndast.6, 23 Til þess að auka NaCl og vatn í berkjuvökva hefur verið reynt að gefa sjúklingum með slímseigjusjúkdóm hýpertónískt (5-7%) saltvatn í innúðaformi. Rannsóknir hafa sýnt að þessi ódýra meðferð reynist vel, bætir lungnastarfsemi og hefur engar langtíma aukaverkanir. Ekki er fullljóst hvenær skal beita þessari meðferð, en líklegt er að hún gagnist flestum sjúklingum.23
Bris
Vanstarfsemi briss hrjáir um 90% sjúklinga með slímseigjusjúkdóm. Hún er talin stafa af stíflum í brisgöngum á grunni gallans í CFTR á svipaðan hátt og lýst er hér að framan. Við þessar aðstæður ná brisensímin ekki að komast inn í meltingarveginn, heldur sitja föst í brisgöngunum þar sem þau virkjast og valda með tímanum eyðingu á vef, bandvefsmyndun og blöðrum.6 Af þessum fíbrótísku blöðrum dregur sjúkdómurinn nafn sitt en þeim var fyrst lýst árið 19361. Afleiðing þessa á meltingu er sú að fita og prótein eru ekki melt sem skyldi og án meðferðar eru sjúklingar með fituskitu og vannærðir.15
Meðferð við brisvanstarfsemi í slímseigjusjúkdómi felst í gjöf brisensíma í töfluformi. Einnig getur vanmelting á fitu leitt til skorts á fituleysanlegum vítamínum (A, D, E og K) og eru sjúklingum því gefin vítamín aukalega.15
Langerhans-frumur í brisinu sem framleiða insúlín verða í fyrstu ekki fyrir merkjanlegum áhrifum vegna bandvefsmyndunar, en með aldri eykst tíðni sykursýki vegna minnkaðrar framleiðslu insúlíns og aukins insúlínviðnáms. Fylgjast þarf reglulega með blóðsykri og meðhöndla sykursýki ef hún greinist en hafa ber í huga að meðferðin er að sumu leyti frábrugðin meðferð sykursýki í öðrum sjúklingum. Skert stjórnun á blóðsykri hefur neikvæð áhrif á lungnastarfsemi og gerir horfur slímseigjusjúklinga almennt verri.15, 37
Meltingarfæri
Gott næringarástand er talið mjög mikilvægt sjúklingum með slímseigjusjúkdóm og það hefur mjög víðtæk áhrif á sjúkdómsganginn.15 Meltingarvökvar eru, eins og seyti annarra kirtla líkamans, þykkari. Fyrir utan að geta leitt til barnabiks-garnastopps (meconium ileus) hjá nýburum, getur seinna á ævinni myndast stífla í smáþörmum (distal ileal obstructive syndrome, DIOS). Reynt er að meðhöndla DIOS með lyfjum en oft þarf að grípa til skurðaðgerða í þeim tilgangi að losa stífluna. Auk þessa er vélindabakflæði algengt og sjúklingar geta með tímanum fengið „fibrosing colopathy“, en það eru bólgur og þrengingar sem myndast í ristli.15
Gallstífla í sjúklingum með slímseigjusjúkdóm getur leitt til gallsteinamyndunar og gall-skorpulifrar (focal biliary cirrhosis), sem síðar getur leitt til portæðar-háþrýstings. Oft gefa lifrar- og gallsjúkdómar engin einkenni, en væg hækkun á lifrarensímum getur verið til staðar.15 Ursodeoxycholic acid (Ursodiol®) er gallsýra sem er oft notuð til meðferðar en gagnsemi lyfsins hefur enn ekki verið að fullu sönnuð með slembirannsóknum.38
Önnur líffæri
Á fósturstigi er þroski sáðrásar mjög háður CFTR próteininu. Þrátt fyrir næga framleiðslu á sæði, eru flestir karlmenn með slímseigjusjúkdóm ófrjóir vegna ófullkomins flutnings á sæði um sáðrásina, og í sumum tilfellum er sáðrásin ekki til staðar frá fæðingu. Hins vegar er oft hægt að nota sæði þeirra til tæknifrjóvgunar. Athyglisvert er að flestir karlmenn sem eru sáðrásarlausir frá fæðingu (CBAVD, congenital bilateral absense of vas deferens) hafa flestir galla í CFTR-geninu sem hefur ekki áhrif á starfsemi annarra líffæra. Þessar stökkbreytingar í CFTR eru það vægar að slímseigjusjúkdómur kemur ekki fram en þær sýna einnig að þroski sáðrásar krefst algerlega eðlilegs CFTR. Til samanburðar má nefna að verulega þarf að draga úr starfsemi CFTR til þess að brissjúkdómur komi fram. Birting slímseigjusjúkdóms í mismunandi líffærum er því háð alvarleika stökkbreytinganna í CFTR.10, 39 Tíðni ófrjósemi hjá konum með slímseigjusjúkdóm er um það bil 20%, en allar eru þó með minnkaða frjósemi sem er tengt vannæringu og óeðlilega þykku slími í leghálsi.15
Sjúklingar með slímseigjusjúkdóm eru almennt með beinþynningu ef miðað er við meðaltalstölur fyrir aldur og kyn. Þessi beinþynning getur leitt til aukinnar hættu á beinbrotum. Kalk í töfluformi hefur verið notað sem meðferð en hefur ekki dugað sem skyldi, og nú er verið að meta hvort bisfosfónöt séu fýsilegur kostur.40
Sjúklingar með slímseigjusjúkdóm geta haft kylfufingur (clubbing) og ofmyndun beins í liðum (hypertrophic osteoarthropathy).41 Í 2-9% tilfella eru til staðar liðbólgur, en þær vara oftast stutt í einu.42 Einnig virðist vera aukin hætta á endurteknum bláæðasegum, en ástæða þess er ókunn.43
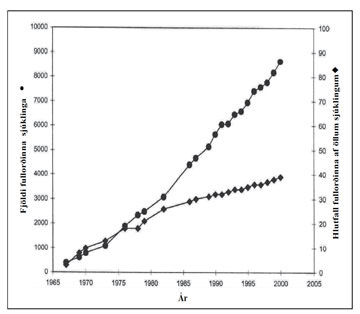
Mynd 1. Sýnir fjölda fullorðinna sjúklinga (eldri en 18 ára) með slímseigjusjúkdóm og hlutfall þeirra af heildarfjölda sjúklinga, á árunum 1965-2001 í Bandaríkjunum.15
Horfur og þróun meðferðar
Það eru einkum eftirfarandi atriði sem gera horfur slímseigjusjúklinga verri en ella: Kvenkyn,
ΔF508 stökkbreyting, sykursýki, vannæring og langvinn berkjusýking með slímmyndandi Pseudomonas aeruginosa. Áður fyrr voru lífslíkur sjúklinga með slímseigjusjúkdóm slæmar og fæstir náðu fullorðinsaldri.6 Horfur hafa batnað umtalsvert, og árið 2005 voru meðallífslíkur sjúklinga í Bandaríkjunum tæp 37 ár.3 Fullorðnum sjúklingum fer þannig stöðugt fjölgandi (mynd 1). og læknar fullorðinna, svo sem lungnalæknar og meltingarlæknar, hafa því þurft að taka æ meiri þátt í meðferð sjúkdómsins í samvinnu við barnalækna.3
Hornsteinar meðferðarinnar eru enn í dag þeir sömu og áður fyrr, góð næring og sýklalyf, en ný sýklalyf vinna betur á sýkingum en þau eldri gerðu. Vaxandi þekking mun væntanlega leiða til nýrra meðferðarmöguleika en í því efni er einkum horft til erfðaefnismeðferðar. Aðrar meðferðarleiðir sem koma til greina eru lyf sem auka seyti klórs og lyf sem minnka seyti natríums. Vonir standa til að yfirstandandi rannsóknir beri árangur og að með nýjum lyfjum líði sjúklingum betur og horfur þeirra vænkist enn frekar.6, 44
Heimildir
1. Baldursson Ó. Function of the Regulatory Domain in the Cystic Fibrosis Transmembrane Conductance Regulator Chloride Channel. Doctoral Dissertation. University of Iceland, Reykjavík 2004.
2. Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245: 1073-80.
3. Cystic Fibrosis Mutation Database. www.genet.sickkids.on.ca/cftr/
4. Welsh MJ, Smith AE. Cystic fibrosis. Sci Am 1995; 273: 52-9.
5. Bergsteinsson H, Baldursson Ó, Clausen M, Cook E, Ólafsson I. Cystic Fibrosis in Iceland 1955?2005; incidence, survival and CFT mutations in the Icelandic population. J Cyst Fibros 2006; 5 Suppl 1:S102.
6. Ratjen F, Döring G. Cystic fibrosis. Lancet 2003; 361: 681-9.
7. Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005; 352: 1992-2001.
8. Preumont V, Hermans MP, Lebecque P, Buysschaert M. Glucose homeostasis and genotype-phenotype interplay in cystic fibrosis patients with CFTR gene deltaF508 mutation. Diabetes Care 2007; 30: 1187-92.
9. Gan KH, Heijerman HGM, Bakker W, et al. Correlation between genotype and phenotype in patients with cystic fibrosis. The Cystic Fibrosis Genotype-Phenotype Consortium. N Engl J Med 1993; 329: 1308-13.
10. Stern RC. The diagnosis of cystic fibrosis. N Engl J Med 1997; 336: 487-91.
11. Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations--correlation with incidence data and application to screening. Hum Mutat 2002; 19: 575-606.
12. Groman JD, Meyer ME, Wilmott RW, Zeitlin PL, Cutting GR. Variant cystic fibrosis phenotypes in the absence of CFTR mutations. N Engl J Med 2002; 347: 401-7.
13. Rodman DM, Polis JM, Heltshe SL, et al. Late diagnosis defines a unique population of long-term survivors of cystic fibrosis. Am J Respir Crit Care Med 2005; 171: 621-6.
14. Wang SS, O?Leary LA, FitzSimmons SC, Khoury MJ. The impact of early cystic fibrosis diagnosis on pulmonary function in children. J Pediatr 2002; 141: 804-10.
15. Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D. Cystic fibrosis adult care: consensus conference report. Chest 2004; 125(1 Suppl):1S-39S.
16. Bals R, Weiner DJ, Wilson JM. The innate immune system in cystic fibrosis lung disease. J Clin Invest 1999; 103: 303.
17. Gibson RL, Burns JL, Ramsey BW. Pathophysiology and Management of Pulmonary Infections in Cystic Fibrosis. Am J Respir Crit Care Med 2003; 168: 918.
18. Brenna AL, Geddes DM. Cystic fibrosis. Curr Opin Infect Dis 2002; 15: 175-82.
19. Cystic fibrosis Europe. www.cfww.org/CFE/ (Accessed Mar 15, 2007).
20. Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 2003; 168:918-51.
21. Hansell DM. Bronchiectasis. Radiol Clin North Am 1998; 36: 107-28.
22. Schneiderman-Walker J, Pollock SL, et al. A randomized controlled trial of a 3-year home exercise program in cystic fibrosis. J Pediatr 2000; 136: 304-10.
23. Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med 2006; 354: 229-40.
24. Fayon M; Airway-Inflammation Group, Societe Francaise de Mucoviscidose. CF-Emerging therapies: Modulation inflammation. Paediatr Respir Rev 2006; 7 Suppl 1:S170-4.
25. Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med 1994; 331: 637-42.
26. Yankaskas JR, Mallory GB. Lung transplantation in cystic fibrosis: consensus conference statement. Chest 1998; 113: 217-26.
27. Rey E, Tréluyer J-M, Pons G. Drug Disposition in Cystic Fibrosis. Clin Pharmacokinet 1998; 35: 313-29.
28. Bolton CE, Ionescu AA, Evans WD, Pettit RJ, Shale DJ. Altered tissue distribution in adults with cystic fibrosis. Thorax 2003; 58: 885-9.
29. Elborn JS, Prescott RJ, Stack BH, et al. Elective versus symptomatic antibiotic treatment in cystic fibrosis patients with chronic Pseudomonas infection of the lungs. Thorax 2000; 55: 355-8.
30. Ramsey BW, Pepe MS, Quan JM, et al. Intermittent Administration of Inhaled Tobramycin in Patients with Cystic Fibrosis. N Engl J Med 1999; 340: 23-30.
31. Bals R, Weiner DJ, Wilson JM. The innate immune system in cystic fibrosis lung disease. J Clin Invest 1999; 103: 303.
32. Eigen H, Rosenstein BJ, FitzSimmons S, Schidlow DV. A multicenter study of alternate-day prednisone therapy in patients with cystic fibrosis. Cystic Fibrosis Foundation Prednisone Trial Group. J Pediatr 1995; 126: 515-23.
33. Lai HC, FitzSimmons SC, Allen DB, et al. Risk of persistent growth impairment after alternate-day prednisone treatment in children with cystic fibrosis. N Engl J Med 2000; 342: 851-9.
34. Nikolaizik WH, Schoni MH. Pilot study to assess the effect of inhaled corticosteroids on lung function in patients with cystic fibrosis. J Pediatr 1996; 128: 271-4.
35. Ásgrímsson V, Guðjónsson T, Guðmundsson GH, Baldursson Ó. Novel effects of Azithromycin on Tight Junction Proteins in Human Airway Epithelia. Antimicrob Agents Chemother 2006; 50: 1805-12.
36. Equi A, Balfour-Lynn IM, Bush A, Rosenthal M. Long term azithromycin in children with cystic fibrosis: a randomised, placebo-controlled crossover trial. Lancet 2002; 360: 978-84.
37. Hardin DS, Moran A. Diabetes mellitus in cystic fibrosis. Endocrinol Metab Clin North Am 1999; 28: 787-800, ix.
38. Cheng K, Ashby D, Smyth R. Ursodeoxycholic acid for cystic fibrosis-related liver disease. Cochrane Database of Systematic Reviews 1999; 3: CD000222. DOI: 10.1002/14651858.
39. Chillon M, Casals T, Mercier B, et al. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med 1995; 332: 1475-80.
40. Conway SP. Bone mineral density in adults with cystic fibrosis. Thorax 1999; 54: 957.
41. Lipnick RN, Glass RB. Bone changes associated with cystic fibrosis. Skeletal Radiol 1992; 21: 115-6.
42. Merkel PA. Rheumatic disease and cystic fibrosis. Arthritis Rheum 1999; 42: 1563-71.
43. Raffini LJ, Raybagkar D, Blumenstein MS, Rubenstein RC, Manno CS. Cystic fibrosis as a risk factor for recurrent venous thrombosis at a pediatric tertiary care hospital. J Pediatr 2006; 148: 659-64.
44. Knowles MR, Hohneker KW, Zhou Z, et al. A controlled study of adenoviral-vector-mediated gene transfer in the nasal epithelium of patients with cystic fibrosis. N Engl J Med 1995; 333: 823-31.