
Fræðigreinar
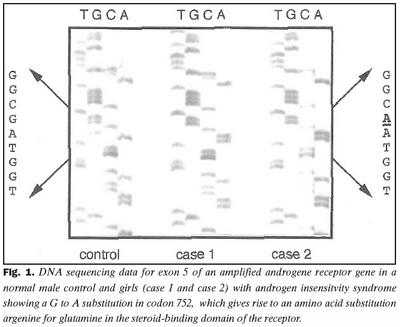
Algjört andrógenónæmi í íslenskri fjölskyldu vegna stökkbreytingar í sterabindistað andrógenviðtækis
Ágrip
Inngangur: Heilkenni andrógenónæmis (androgen insensitivity syndrome, AIS) er sjúkdómur, sem erfist kynbundið víkjandi, og hefur í för með sér truflun á eðlilegri kynþróun hjá karlfóstri, sem á sér stað fyrir tilstilli testósteróns. Orsök heilkennisins er í flestum tilvikum stökkbreyting í geni andrógenviðtækis á X-litningi. Í þessari rannsókn er íslenskri fjölskyldu lýst, þar sem fundist hafa tvær stúlkur með heilkenni andrógenónæmis. Leitað var að stökkbreytingum í geni andrógenviðtækisins í fjölskyldunni.Efniviður og aðferðir: Erfðaefni var einangað úr blóði tveggja stúlkna með algjört andrógenónæmi svo og nánustu ættingja þeirra. Fjölliðunarhvarf var notað til að fjölfalda allar átta útraðirnar í geni andrógenviðtækis tilfella með AIS. SSCP-aðferðin var notuð til að skima fyrir stökkbreytingum í geninu. Niturbasaröð þeirrar útraðar sem gaf óeðlilegt SSCP mynstur var síðan ákvörðuð. Greiningaraðferð, sem byggir á fjölliðunarhvarfi og skerðibútabreytileika, var þróuð og notuð til að finna stökkbreytta samsætu meðal fjölskyldumeðlima.
Niðurstöður og ályktun: Með notkun SSCP og niturbasaraðgreiningar fannst sama stökkbreytingin í útröð 5 í geni andrógenviðtækis stúlknanna tveggja með AIS. Niturbasaröðin CGA var stökkbreytt í CAA, sem hefur í för með sér að í stað amínósýrunnar argeníns í stöðu 752 kemur glútamín (R752Q samsæta). Þessi stökkbreyting er staðsett í sterabindistað andrógenviðtækisins og hefur í för með sér að það getur ekki bundið testósterón.
Niðurstöður rannsókna á fjölskyldumeðlimum með sérstakri erfðagreiningaraðferð fyrir samsætuna R752Q sýndu að um var að ræða nýja (de novo) stökkbreytingu í kynfrumum móðurömmu, þar sem ekki var hægt að greina samsætuna í erfðaefni hvítra blóðkorna hennar. Erfðagreiningin skapaði grundvöll til erfðaráðgjafar hjá fjölskyldunni.
English Summary |
| Ólafsson Í, Kristjánsson K, Hjaltadóttir G, Schwartz M, Þórsson ÁV Complete androgen insensitivity in an Icelandic family caused by mutation in the steroid binding region of the androgen receptor Læknablaðið 2000; 86: 163-6 Introduction: Androgen insensitivity syndrome (AIS) is a X-linked rescessive disorder characterized by impairment of the androgen-dependant male sexual differentiation. The cause of AIS is in most cases a mutation in the gene of the androgen receptor on the X chromosome. In this study we describe an Icelandic family with two girls with AIS. A search for mutations in the androgen receptor gene was performed in order to identify the genetical and molecular basis for AIS in this family. Material and methods: Genomic DNA was isolated from two girls with complete AIS and their close relatives. PCR was used to amplify all eight exons of the androgen receptor gene of the two AIS girls and SSCP used to screen for mutations. DNA fragments showing abnormal SSCP pattern were subjected to nucleotide sequencing. PCR based diagnostic method was developed and used to detect the mutation causing AIS in the family. Results and conclusions: Using SSCP and DNA sequencing a CGA to CAA missense mutation in exon 5 at codon 752 was identified. The mutation causes in an Arg to Gln amino acid substitution (R752Q mutation) in the ligand binding domain of the androgen receptor and a complete androgen insensitivity. Members of the family were genotyped using a PCR based method for identification of the mutant allele. The results strongly indicated a de novo mutation in a germ cell of the maternal grandmother, as the mutation was not found in her blood leucocytes. The diagnostic test provided a basis for genetic counselling for the family. Key words: sex differentiation, testosterone, androgen receptor, mutation. Correspondence: Ísleifur Ólafsson. E-mail: isleifur@shr.is |
Inngangur
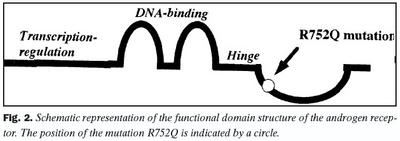
Eðlileg þróun á kynfærum karla á fósturstigi er afar flókið líffræðilegt ferli. Mikilvægur þáttur í því ferli er myndun andrógenstera og tenging þeirra við viðtæki sitt. Ef andrógensterar myndast ekki eða andrógenviðtæki starfa ekki eðlilega truflast kynþróunin. Slík truflun veldur klínískri svipgerð sem kölluð er heilkenni andrógenónæmis (androgen insensitivity syndrome, AIS) (1,2). Heilkennið er í langflestum tilvikum orsakað af stökkbreytingu í geni andrógenviðtækis, en í einstaka tilvikum getur skortur á ensýminu 5a-redúktasa valdið klínískt svipuðu heilkenni (1).Gen andrógenviðtækis er staðsett á X-litningi og erfist AIS kynbundið víkjandi (3). Genið er samsett út átta útröðum og nær yfir 90 kb bút af litningnum (4,5). Það er afritað í flestum vefjum líkamans, en stjórnun á tjáningu þess virðist afar flókin (6). Sjálft andrógenviðtækið er samsett úr 919 amínósýrum, vegur 110-114 kDa og tilheyrir hópi þróunarfræðilega skyldra prótína sem kallaður hefur verið kjarnaviðtækjafjölskyldan (nuclear receptor superfamily) (7-11). Af þeim fjölda prótína sem tilheyra þessari fjölskyldu má nefna sykurstera-, estrógen-, D-vítamín-, A-vítamín- og skjalkirtilshormónviðtækið. Skipta má andrógenviðtækinu og skyldum prótínum upp í fjóra byggingarhluta (12,13). Þeir eru: 1) amínóendi sem ber þann hluta viðtækisins sem miðlar örvun á genaafskrift, 2) DNA bindisvæði sem inniheldur tvo svokallaða sínkfingur og þekkir sérstakar raðir í prómóter svæði gena, 3) hjararhluti sem ber markröð fyrir frumukjarna (nuclear targeting signal) og 4) sterabindistaður sem bindur andrógen. Þegar andrógensteri hefur bundist viðtæki sínu í umfrymi verða breytingar í byggingu viðtækisins, sem valda því að það berst inn í kjarna frumunnar. Þar binst viðtakinn ákveðnum niturbasaröðum í prómóter svæði útvalinna gena og hefur áhrif á tjáningu þeirra.
Í gagnagrunni yfir allar stökkbreytingar, sem hafa fundist í andrógenviðtækinu (netfang http://www. mcgill.ca/androgendb/) er lýst rúmlega 300 mismunandi stökkbreytingum (14). Þessar stökkbreytingar eru af ýmsum gerðum og valda mismiklum klínískum einkennum eftir því hve mikil áhrif þær hafa á lífefnafræðilega virkni viðtækisins (1,2). Í þessari rannsókn er íslenskri fjölskyldu lýst, þar sem fundist hafa tvær stúlkur með litningagerðina 46, XY og klínísk einkenni algjörs andrógenónæmis. Gerð var leit að stökkbreytingum í geni andrógenviðtækis stúlknanna og nánustu ættingjar voru einnig rannsakaðir.
Efniviður og aðferðir
Úrtak: Blóðsýni voru tekin úr tveimur systradætrum sem höfðu litningagerðina 46, XY og höfðu greinst með klínísk einkenni andrógenónæmis. Sýni voru einnig tekin úr mæðrum þeirra, mæðrasystrum, móðurömmu og ömmusystrum. Erfðaefnið var einangrað úr hvítum blóðkornum eins og áður hefur verið lýst (15). Rannsóknin var gerð með upplýstu samþykki allra þátttakenda. Vísindasiðanefnd Sjúkrahúss Reykjavíkur og Tölvunefnd gerðu ekki athugasemd við birtingu niðurstaðna.Fjölliðunarhvörf: Gerð var leit að stökkbreytingum í geni andrógenviðtækis hjá stúlkunum tveimur með AIS. Allar átta útraðirnar í geni andrógenviðtækisins voru fjölfaldaðar með því að nota 14 pör fákirna, en niturbasaröð þeirra hefur áður verið lýst (16). Annað fákirnanna í þessum 14 pörum var merkt með bíótíni í 5'-enda.
SSCP rannsókn: Svokölluð SSCP aðferð (single-stranded conformation polymorphism) var notuð til að finna stökkbreytingu í fjölfölduðum DNA bútum úr geni andrógenviðtækisins (16,17). Bútarnir voru rannsakaðir með því að blanda þremur mL af hverri fjölliðunarblöndu við þrjá mL af hleðslustuðpúða (95% formamíð, 20 mM EDTA, 0,05% brómófenól blátt og 0,05% xýlen cýanól). Blandan var hituð við 100°C í fimm mínútur og síðan snöggkæld á ís. Tveimur mL af sýnunum var hlaðið á 20% einsleitt PHAST pólýakrílamíð gel (Amersham Pharmacia Biotech). Fyrir hleðslu sýna voru gelin undirbúin með 10 meðalvolttímum við 5 mA, 1 W og 400 V. Sýnin voru fyrst rafdregin í tvo meðalvolttíma við 5 mA, 1 W og 25 V og síðan 450 meðalvolttíma við 5 mA, 1 W, og 400 V. Eftir rafdrátt voru DNA bútar í polýakrílamíð geli gerðir sýnilegir með silfurlitun.
DNA niturbasaraðgreining: Sú útröð andrógenviðtækis, sem gaf óeðlilegt SSCP mynstur við rafdrátt (niðurstöður ekki sýndar), var rannsökuð frekar með niturbasaraðgreiningu. Fyrir raðgreininguna voru DNA-bútar hreinsaðir úr fjölliðunarblöndum með QIAquick PCR Purification Kit frá Qiagen. Einstrengja DNA-bútar voru síðan einangraðir með notkun á Dynabeads M-250 streptavidín (Dynal) og Dynal segli. Þessir bútar voru síðan raðgreindir samkvæmt aðferð Sangers með SequenaseTM Kit útgáfa 2.0 (USB), [a-35S]-dATP og þeim fákirnum, sem notuð voru til að fjölfalda viðkomandi útröð.
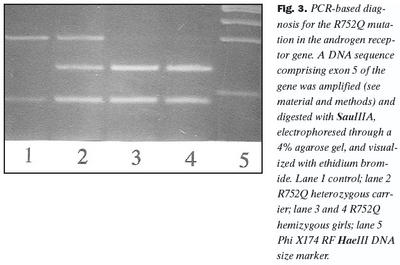
Arfgerðargreining: Sérstök aðferð til arfgerðargreiningar á R752Q samsætunni var þróuð, sem byggir á fjölliðunarhvarfi og skerðibútabreytileika. Tvö hundruð og níutíu basapara (bp) kjarnsýrubútur, sem inniheldur útröð 5 í geni viðtækisins, var fjölfaldaður í fjölliðunarhvarfi með hjálp fákirnanna 5´-CAACCCGTCAGTAGTACCCAGACTGACC-3´ og 5´-AGCTTCACTGTCACCCCATCACCATC-3´ og voru 40 hringir endurteknir við hitastigin 93°C, 63°C og 72°C. Tveimur einingum af skerðiensíminu SauIIIA var síðan blandað við 10 mL af fjölliðunarblöndu og skerðibútabreytileiki greindur með rafdrætti í 4% agarósu geli. Rannsókn á einstaklingum með eðlilega samsætu andrógenviðtækis gefur skerðibúta sem eru 139, 107 og 44 bp að lengd, á einstaklingum, sem eru arfblendnir með tilliti til R752Q samsætunnar, 183, 139, 107 og 44 bp; og hjá arfstökum (hemizygous) einstaklingum með tilliti til R752Q samætunnar, 183 og 107 bp.
Niðurstöður
Við fjölföldun og SSCP rannsókn á útröðum í geni andrógenviðtækis kom fram erfðabreytileiki í útröð 5 hjá stúlkunum tveimur með AIS (niðurstöður ekki sýndar). Aðrar útraðir gensins gáfu ekki merki um breytileika. Niturbasaröð útraðar 5 í geni andrógenviðtækis stúlknanna var ákvörðuð og fundust basaskiptastökkbreyting í amínósýrukóða 752 (mynd 1). Þar sem eðlilegur einstaklingur hefur röðina CGA var röðin CAA hjá stúlkunum. Samkvæmt erfðalykli (genetic code) hafa þessi basaskipti í för með sér að í stað argeníns í stöðu 752 í fjölpeptíðkeðju andrógenviðtækisins kemur amínósýran glútamín (R752Q). Þessi amínósýra er staðsett í sterabindistað viðtækisins (mynd 2). Stökkbreytingunni R752Q í andrógenviðtækinu hefur einu sinni áður verið lýst, en það var hjá tveimur náskyldum stúlkum, sem einnig höfðu klínísk einkenni algjörs andrógenónæmis (2,18). Rannsóknir á andrógenbindigetu ræktaðra fíbróblasta frá kynfærum þessara stúlkna sýndu ekki mælanlega bindingu andrógens.Einföld aðferð til að greina R752Q stökkbreytinguna var þróuð, en hún byggðist á fjölliðunarhvarfi og skerðibútabreytileika. Niturbasaskiptin í kóða 752 valda því að skerðistaður fyrir skerðiensímið SauIIIA hverfur og var það notað við að finna R752Q samsætuna. Niðurstöður við rannsókn á heilbrigðum, R752Q arfblendnum og R752Q arfstökum einstaklingum er sýnd á mynd 3.
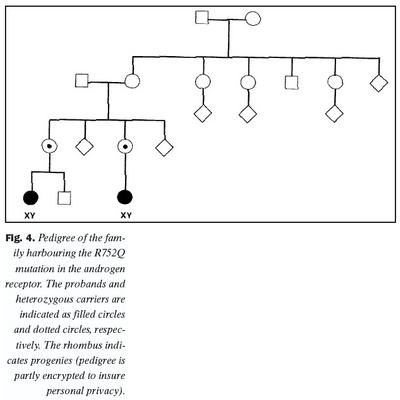
Aðferð til arfgerðargreiningar á samsætunni R752Q var notuð til að finna arfbera innan fjölskyldunnar. Báðar stúlkurnar með litningagerðina 46, XY og klínísk einkenni AIS reyndust arfstakar fyrir stökkbreytingunni (mynd 4). Báðar mæður þeirra báru stökkbreytinguna á arfblendnu formi, en mæðrasystur voru allar eðlilegar. Stökkbreytinguna var hins vegar ekki að finna í hvítum blóðkornum móðurömmu stúlknanna, né heldur systrum móðurömmu. Þessar niðurstöður benda eindregið til þess að um sé að ræða nýja stökkbreytingu (de novo) í gen andrógenviðtækis í kynfrumum móðurömmu. Mæður stúlknanna með AIS hafa erft stökkbreyttu samsætuna frá móðurömmu, en þar sem um er að ræða kynbundnar víkjandi erfðir, birtist klínísk svipgerð samsætunnar ekki í þeim.
Umræða
Í þessari rannsókn var aðferðum erfðatækninnar beitt til þess að finna stökkbreytingu í geni andrógenviðtækis hjá stórri fjölskyldu með tvær stúlkur með litningagerðina XY og klínísk einkenni algjörs andrógenónæmis. Stökkbreytingin fólst í niturbasaskiptum í kóða 752 í fjölpeptíðkeðju andrógenviðtækisins og hefur í för með sér að glútamín kemur í stað argeníns í sterabindistað viðtækisins (R752Q). Þessi stökkbreyting er fyrsta stökkbreytingin í andrógenviðtækinu sem lýst er í íslenskri fjölkyldu en á heimsvísu hefur yfir 300 stökkbreytingum basaskiptastökkbreytingum, brottföllum og innskotum verið lýst í geninu. Hér á landi er vitað um nokkur fleiri tilfelli af AIS en erfðafræðileg orsök þeirra hefur ekki fundist.Erfðagreining á nánustu skyldmennum stúlknanna með AIS sýndi R752Q samsætuna hjá mæðrum þeirra, en endurteknar rannsóknir á erfðaefni úr hvítum blóðkornum móðurömmu sýndu eðlilega arfgerð. Sú niðurstaða bendir eindregið til að ný stökkbreyting hafi orðið í kynfrumum hennar. Í nýlegri rannsókn á fjölskyldum með aðeins einum einstaklingi með AIS reyndust átta af 30 (27%) bera nýjar stökkbreytingar (19). Í fimm tilfellum af þessum átta var um að ræða stökkbreytingu í kynfrumu móður, en í þremur reyndust tilfellin vera með mósaík arfgerð. Hjá 22 (73%) fjölskyldum voru mæður arfblendnar. Þessar niðurstöður sýna háa tíðni í myndun nýrra stökkbreytinga í geni andrógenviðtækisins og styðja einnig tilgátu Haldanes um að nýjar stökkbreytingar á X-litningi þurfi til þess að viðhalda arfgengum kynbundnum sjúkdómum, sem minnka lífslíkur og frjósemi arfbera (20,21).
Ný þekking og nýjar aðferðir í erfðafræði hafa gert læknum mögulegt að greina með mikilli nákvæmni orsakir ættlægra sjúkdóma. Í þeirri stóru fjölskyldu sem hér er lýst voru þessar aðferðir notaðar til greiningar á arfberum AIS. Þannig var með öruggum hætti hægt að veita fjölskyldunni erfðaráðgjöf og stór hluti hennar útilokaður sem hugsanlegir arfberar.
Þakkir
Vísindasjóður Sjúkrahúss Reykjavíkur fær þakkir fyrir fjárhagslegan stuðning.Heimildir
1. Griffin JE. Androgen resistance - The clinical and molecular spectrum. New Engl J Med 1992; 326: 611-8.2. Quigley CA, De Bellis A, Marschke KB, El-Awady, Wilson EM, French FS. Androgen receptor defects: historical, clinical and molecular perspectives. Endocr Rev 1995; 16: 271-321.
3. Lubahn DB, Joseph DR, Sullivan PM, Willard HF, French FS, Wilson EM. Cloning of human androgen receptor complementary DNA and localization to the X chromosome. Science 1988; 240: 327-30.
4. Lubahn DB, Brown TR, Simental JA, Higgs HN, Migeon CJ, Wilson EM, et al. Sequence of the intron/exon junctions of the coding region of the human androgen receptor gene and identification of a point mutation in a family with complete androgen insensitivity. Proc Natl Acad Sci USA 1989; 86: 9534-8.
5. Kuiper GGJM, Faber PW, van Rooij HJC, Van der Korput JAGM, Ris-Stalpers C, Klaassen P, et al. Structural organization of the human androgen receptor gene. J Mol Endocrineol 1989; 2: R1-R4.
6. Wolf DA, Herzinger T, Hermeking H, Blaschke D, Horz W. Transcriptional and posttranscriptional regulation of human androgen receptor expression by androgen. Mol Endocrinol 1993: 7; 924-36.
7. Lubahn DB, Joseph DR, Sar M, Tan J-A, Higgs HN, Larsson RE, et al. The human androgen receptor: complementary deoxyribonucleic acid cloning, sequence analysis and gene expression in prostate. Mol Endocrinol 1988; 2: 1265-75.
8. Chang C, Kokontis J, Liao S. Structural analysis of complementary DNA and amino acid sequences of human and rat androgen receptors. Proc Natl Acad Sci USA 1988; 85: 7211-5.
9. Trapman J, Klaassen P, Kuiuper GGJM, Van der Korput JAGM, Faber PW, van Rooij HJC, et al. Cloning, structure and expression of a cDNA encoding the human androgen receptor. Biochem Biophys Res Commun 1988; 153: 241-8.
10. Martinez E, Moore DD, Keller E, Pearce D, Heuvel JPV, Robinson V, et al. The nuclear receptor resource: a growing family. Nucl Acid Res 1998; 26: 239-41.
11. Beato M, Herrlich P, Schütz G. Steroid hormone receptors: many actors in search of a plot. Cell 1995; 83: 851-7.
12. Jenster G, Van der Korput JAGM, van Vroonhoven C, van der Kwast TH, Trapman J, Brinkman AO. Domains of the human androgen receptor involved in steroid binding, transcriptional activation and sucellular localization. Mol Endocrinol 1991; 5: 1396-404.
13. Simental JA, Sar M, Lane MV, French FS, Wilson EM. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Biol Chem 1991; 266: 510-8.
14. Gottlieb B, Lehvaslaiho H, Breitel LK, Lumbroso R, Pinsky L, Trifiro M. The androgen receptor gene mutations database. Nucl Acid Res 1998; 26: 234-8.
15. Bell GI, Karam JH, Rutter WJ. Polymorphic DNA region adjacent to the 5' end of the human insulin gene. Proc Natl Acad Sci USA 1981; 78: 5759-63.
16. Batch JA, Wiliams DM, Davies HR, Brown BD, Evans BAJ, Hughs IA, et al. Androgen receptor gene mutations identified by SSCP in fourteeen subjects with androgen insensitivity syndrome. Hum Mol Gen 1992; 1: 497-503.
17. Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T. Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci USA 1989; 86: 2766-70.
18. Brown TR, Newmark J, Ghirri P. Six naturally occurring mutations in the 144-base-pairs of exon E of the human androgen receptor gene. Program of the 74th Annual Meeting of The Endocrine Society, San Antonio, TX, 1992, p 428 [abstract 1506].
19. Hiort O, Sinnecker GHG, Holterhus P-P, Nitsche EM, Kruse K. Inherited and de novo androgen receptor gene mutations: Investigation of single-case families. J Pediatr 1998; 131: 939-43.
20. Leslie ND. Haldane was right: de novo mutations in androgen insensitivity syndrome. J Pediatr 1998; 131: 917-8.
21. Haldane JBS. The rate of spontaneous mutation of a human gene. J Genet 1935; 35: 317-26.